
摘要:本教程演示了Flare的对接计算流程,包括:1)PDB文件下载;2)受体、配体结构准备;3)配体的导入;4)分子对接计算;5)结果分析与可视化。
一. 背景介绍
BCR-ABL激酶是抗白血病药物伊马替尼(Imatinib)的作用靶点,复合物晶体结构的PDB代码为1IEP。本教程将从伊马替尼的SMILES代码出发,进行对接计算,以演示如何用FLARE进行分子对接、结合模式预测。
Figure 1. 伊马替尼的结构
Flare采用分子对接软件Lead Finder作为计算引擎, 在同样的数据集上Lead Finder展示出比同行们更好的分子对接计算精度(见表1)。借助于Flare优秀的分子结构准备功能,使得Flare版的Lead Finder的性能优于Lead Finder。
Table 1. Lead Finder的对接计算精度(redocking)
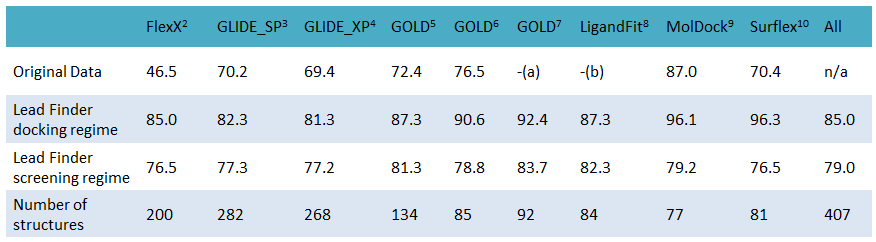
数据源自: http://biomoltech.com/science/benchmarking/docking_success_rate
Flare/leadfinder分子对接技术流程如下图所示:
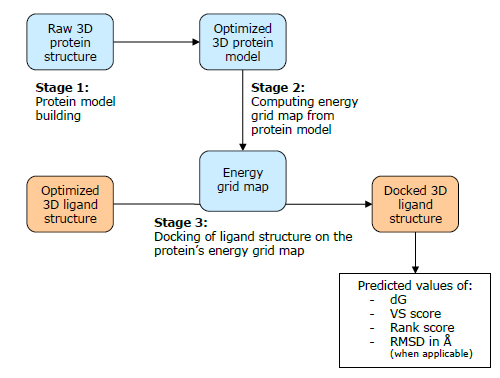
其中,第一步是从原始蛋白3D结构出发得到优化的蛋白模型,是由LeadFinder的build_model模块在后台完成,对应于第三节的蛋白结构准备。第二步energy grid map生成是由Leadfinder完成,第三节的参数设定中的grid define就是定义结合位点(结合口袋)的位置以计算格点。格点文件包含了蛋白结合位点的全部信息,在分子对接计算中就是用格点代表蛋白。格点文件可以重复使用,对接不同的配体到同一个结合位点就可以用同一个格点文件。有了格点与准备好的配体结构,就可以开始分子对接计算。分子对接是将指定的配体对接到代表蛋白的格点上,并给出pose与打分值。
二. 准备工作
- 安装了Flare
- 电脑可以上网,以便下载PDB文件(1IEP)
- 准备好伊马替尼的结构文件,比如SMILES、Mol、SDF或Mol2格式文件。你也可以从这里下载一个SMILES格式文件:imatinib.smi
化合物结构准备,或化合物库准备方法可以参考:http://blog.molcalx.com.cn/2017/06/05/tutorial-chemistry-library.html
三. 分子对接计算
- 下载PDB文件,准备蛋白结构
- 导入待对接的化合物到Ligand表单中
- 分子对接
(1)File | Download PDB
(2)在PDB codes行键入1IEP,点击OK按钮,开始下载PDB结构。
下载完毕,会弹出Open Molecules对话框,确保Protonation state行为Do full preparation on proteins and ligands,点击Open按钮,会弹出Flare protein preparation对话框。

Figure 2. Flare protein preparation
在Cap Chains, Auto-extract ligand打钩;并将Active site size设置为6埃。然后点击Start按钮开始准备蛋白结构。
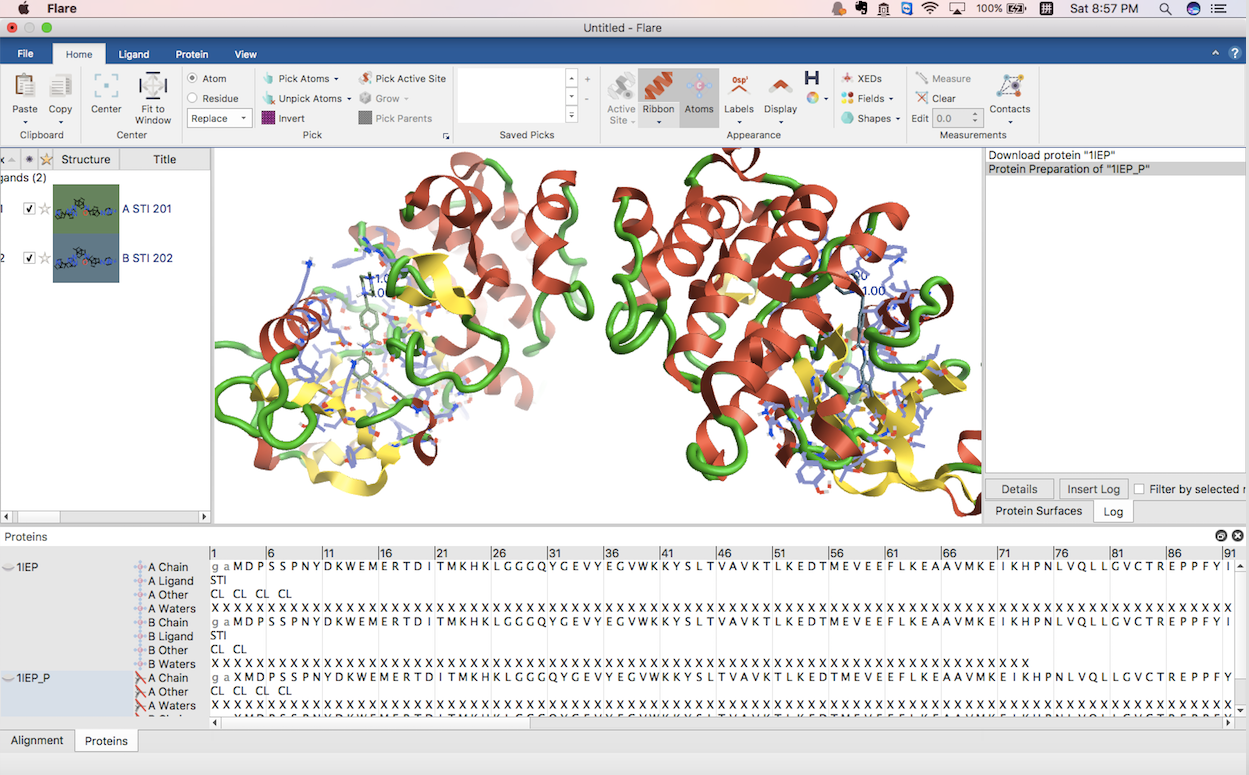
Figure 3. Flare结构准备结束。在本例中,ligand表单含有两个配体,分别来自Chain A的A STI 201与来自Chain B的B STI 202;Protein表单含有套结构一个为1IEP,另一个为1IEP_P。其中1IEP为原始下载的结构文件,而1IEP_P为Flare准备好的文件。
(3)表单的编辑:删除Chain B的小分子、蛋白、水以及其他。
按住Shift键,在Protein表单的1IEP_P下用鼠标分别点击B Chain,B Other与B Waters,则选中蛋白B链的全部内容;单击鼠标右键,选Delete,删除选中的内容。
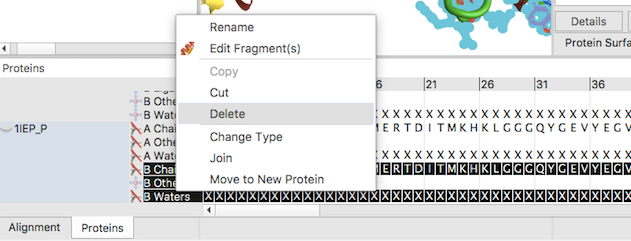
Figure 4. 删除Chain B的内容:蛋白,其他以及水
采用类似的操作,可以删除Ligand表单中的配体B STI 202,Chain A蛋白中的四个Cl离子。
注意:本文只是一个演示,在具体实践中要删除什么、保留什么需要根据您具体的研究内容而定。
File | Open 读入imatinib.smi文件,点击Open,会弹出Open Molecules对话框。对话框的Molecule Type设置为Ligands;其它部分采用默认值,点击Open。结构准备完毕,在Ligand表单会新增一个分子Imatinib/Gleevec,我们要将这个分子docking到蛋白口袋里。
(1) 打开对接对话框
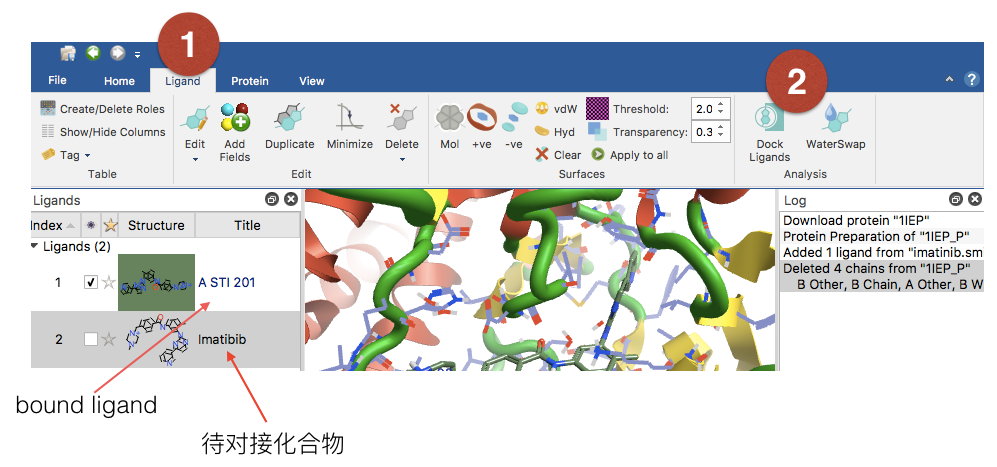
Figure 5. Flare分子对接
在Ligand表单里点击待对接化合物,再点击Ligand菜单里的Dock Ligands命令(Figure 5)打开分子对接计算对话框,见Figure 6。
(2)参数设定
这部分分为两个子任务:生成分子对接的格点文件与分子对接计算。分子对接格点需要定义对接计算的结合位点在哪里,有三种方式:从结合位点里选一个原子来指示结合位点;用结合的配体指示结合位点;用计算现成的格点。
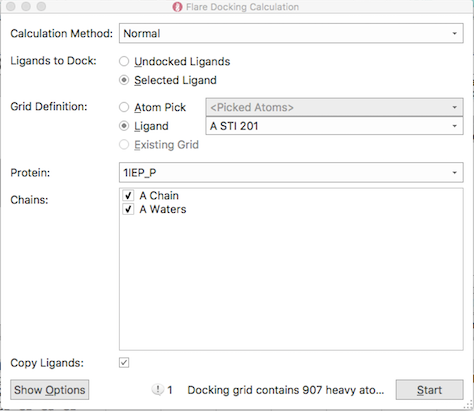
Figure 6. Flare分子对接参数设定
在Flare docking calculation对话框里,设置参数如下:
Calculation method:Normal
Calculation method设置计算模式:quick,normal,accurate but slow, score only。根据不同的目的选择不同的模式。
Ligands to dock: selected ligands
Undocked ligands: 所有未对接过的分子都会被进行计算;Selected ligands:仅是那些被选中的化合物会被进行对接计算。
Grid Definition: Ligand | A STI 201
本计算中我们用共晶的配体(bound ligand)来定义结合位点、计算格点。Lead Finder用预先生成的GRID文件对pose进行打分,因此需要在对接前准备好GRID文件。准备GRID文件最重要的一点是定义分子对接的结合位点在哪里,可以通过复合物结构里的配体来定义(ligand),还可以选择一个或多个原子来定义(选择Atom pick,然后再3D视窗区用鼠标选择活性位点内的一个原子或几个原子)。
Protein: 1IEP_P
并将A Chain与A Waters打钩,意思是蛋白与水都设为蛋白。如果不想把水看作为蛋白的一部分,则将water处不要打勾。注意:本文只是演示Flare对接的功能与选项的作用,并不是推荐默认将水设定为蛋白的一部分。
其它选项:Show options
一般情况下,不需要对show options进行设置,采用默认的预设值就可以。但是,有时候准备的配体初始结构不理想,则需要通过show options的设置来提高计算精度。比如,对化合物进行优化;允许所有的酰胺键、酯键、共轭单键进行旋转。
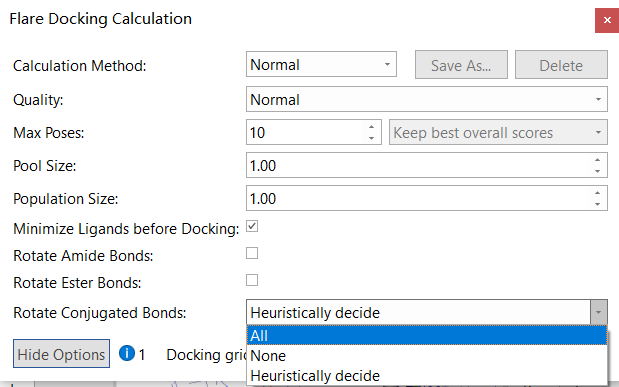
一般情况下,酰胺键是反式的,所以不对酰胺键进行旋转。如果不能确认初始的构象是否合理,可以全部打钩。注意,最后一项的共轭键是指共轭单键(比如甲氧基苯的的氧原子与苯环的共轭),如果你对初始构象不了解,建议选All。尤其是ChemOffice的用户画的酰胺3D结构经常有顺势结构,所以建议ChemOffce/Chem3D用户将所有可选项都打钩、并且共轭键旋转选择All。
(3)开始分子对接计算
点击Start按钮,开始计算。分子对接计算分两个子任务:格点计算与分子对接。格点计算耗时较长,但无需重复计算。计算结束会在ligand表单出现一个新的分子,其名称为待对接化合物加上copy。
四.对接结果可视化与分析
对接的结果保存在配体表单里。
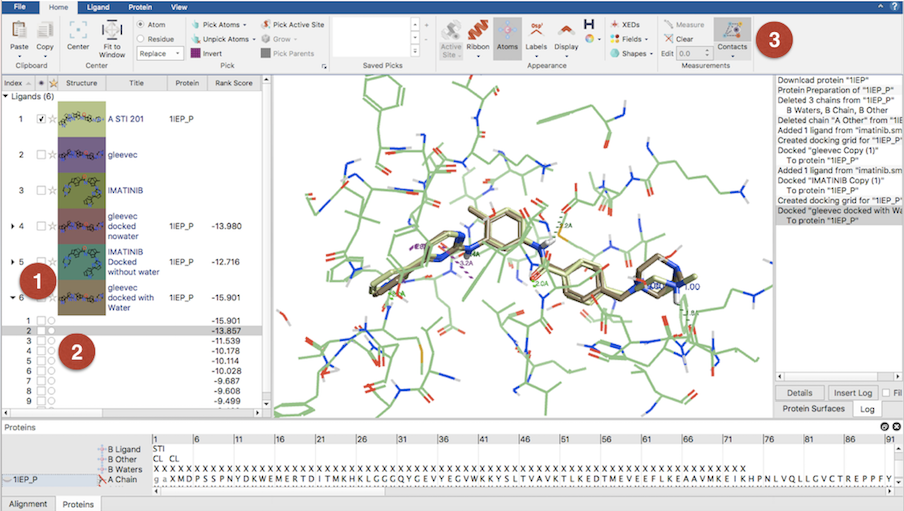
Figure 7. 对接结果的可视化与分析
1. Pose的操作
点击任意一个对接后配体左边的小箭头(Figure 7 图标1)可以展示保存的pose。通过键盘的上、下键可以滚动浏览对接的pose,也可以用鼠标选中pose。点击单选按钮(Figure 7 图标2)可以收藏喜欢的pose。
Ligand表单里的Protein列给出当前docking计算是跟哪个蛋白进行的,将pose与蛋白关联起来。
2. 配体-蛋白相互作用的展示
首先确认在Protein窗口选中对接时用的蛋白,再点击Contacts(Figure 7, 图标3),可以展示蛋白与pose之间的相互作用:氢键、pi-pi相互做用等等(Figure 8)。
Figure 8. Contacts展示配体-蛋白相互作用类型
3. 关于对接结果的导出
对接的pose可以导出, 使用菜单: File | Export | Export Selected ligands。支持的格式包括: SDF,Mol2, PDB, XED。
4. 关于打分值
- Rank score
- dG score
- VSscore
每个配体对接获得的pose按Rank Score值从小到大排序,也即打分最佳的靠前,Rank Score设计的意图是让对方最佳的pose最接近实验结果。
dG打分函数设计用来进行精确的评估蛋白-配体结合自由能。
VSscore(Virtual Screening scoring)打分:该打分设计用于虚拟筛选,高效率的区分活性(高打分->true binders)与非活性化合物(打分低->Inactive ligands)。
五. 接下来可以做什么
- 用WaterSWAP对pose进行精确打分、排序;
- 用Protein Field Surface观察配体与蛋白在静电场上的关系,设计新的分子;
- 用静电电互补性考察分子的结构,提出结构改造方案;
- 用Ligandscout生成基于结构的药效团;
- 计算水分子的位置与稳定性,设计新的分子;
- 多个分子对接后,用AutoT&T进行全新分子设计。


