
摘要:Flare是一款开创性的蛋白-配体分析与设计软件,它整合了当今最尖端的开源与商业技术,为基于结构的设计带来了新视角。Flare包含了:1)结构准备;2)蛋白静电相互作用势分析;3)热力学积分法研究配体-蛋白的相互作用能;4)水分子计算;5)分子对接等功能。当前版本为Beta版,现在开放测试。
尊敬的老师,同学:
Flare是一款开创性的蛋白-配体分析与设计软件,它整合了当今最尖端的开源与商业技术,为基于结构的设计带来了新视角。我们很高兴的通知您,目前Flare已经开发到Beta版本,接受开放测试。请有兴趣的老师、同学联系我们进行测试。我们为您提供免费的测试版软件,您需要在测试之后给我们反馈结果与意见。
祝您工作顺利!
广州市墨灵格信息科技有限公司
2017年4月21日
一. 用Flare您可以:
- 简便的蛋白-配体复合物结构导入(下载)
- 精确可靠的蛋白结构准备
- 序列比对与结构叠合
- 蛋白相互作用势分析
- 精确的分子对接(结合模式预测)
- 蛋白-配体复合物结构优化
- 3D-RISM分析:计算水分子在结合位点里的稳定性与位置(用Cresset XED与AMBER力场)
- WaterSWAP分析:研究蛋白-配体相互作用,计算蛋白-配体结合自由能

“蛋白相互作用势可以凸显出不同家族靶标的公共特征,借助这些信息,为分子设计提供了前所未有的设计方向”。
二. 蛋白相互作用势:让您的分子设计完善
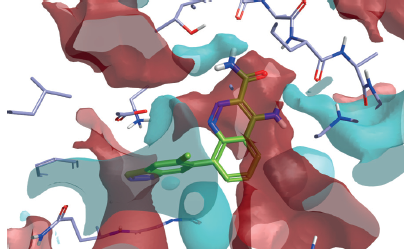
蛋白相互作用势
Flare用XED力场计算蛋白活性位点的静电特性。相互作用势可以告诉您配体-蛋白结合的关键知识,帮助您完善分子的设计。用蛋白相互作用势可以:
- 揭示结合位点的静电特征
- 理解配体结合的要求
- 比较配体与蛋白的静电
- 改进配体的设计
三. WaterSWAP: 计算配体-蛋白的结合自由能
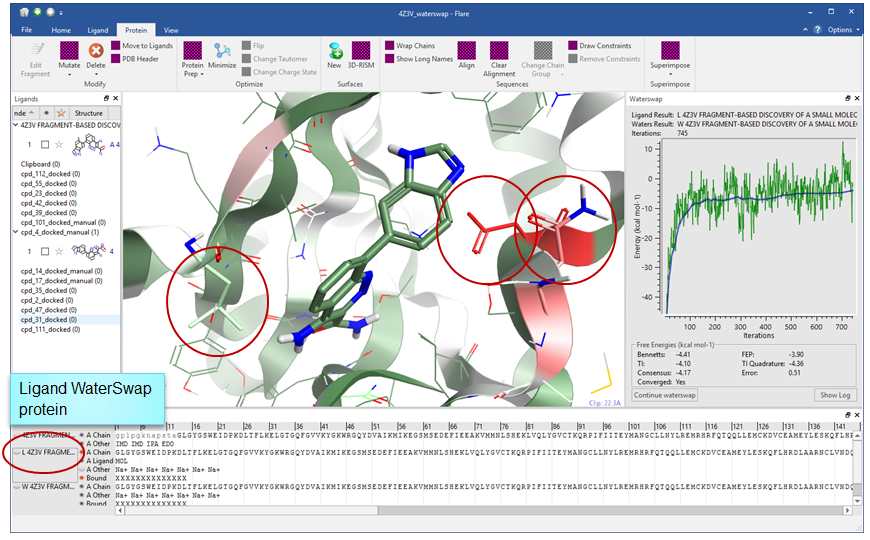
WaterSWAP
WaterSwap:计算配体-蛋白的绝对结合自由能。它可以用于:
- 研究配体结合的能量学特点
- 将结合能分解为每个残基的贡献以发现优选的配体相互作用模式
- 计算一系列化合物的结合自由能(ΔG of binding)以帮助您优选配体
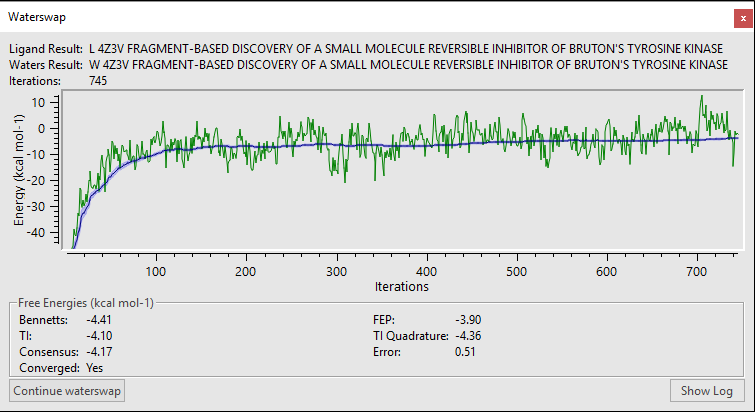
WaterSWAP的计算结果:上图的曲线展示了每次迭代的能量计算结果;下面的方框展示了配体-蛋白的几种不同计算方法的结合自由能(Bennett, TI, FEP, TI Quadrature)。
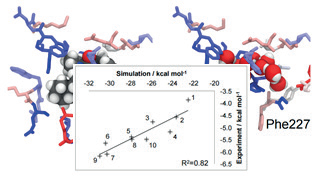
WaterSWAP的计算结果:结合位点里的残基具有不同的着色;红色代表着其偏向于与水发生相互作用;绿色意味着偏向于与配体发生相互作用;白色意味着没有偏好。
四. 3D-RISM: 计算水的稳定性与位置
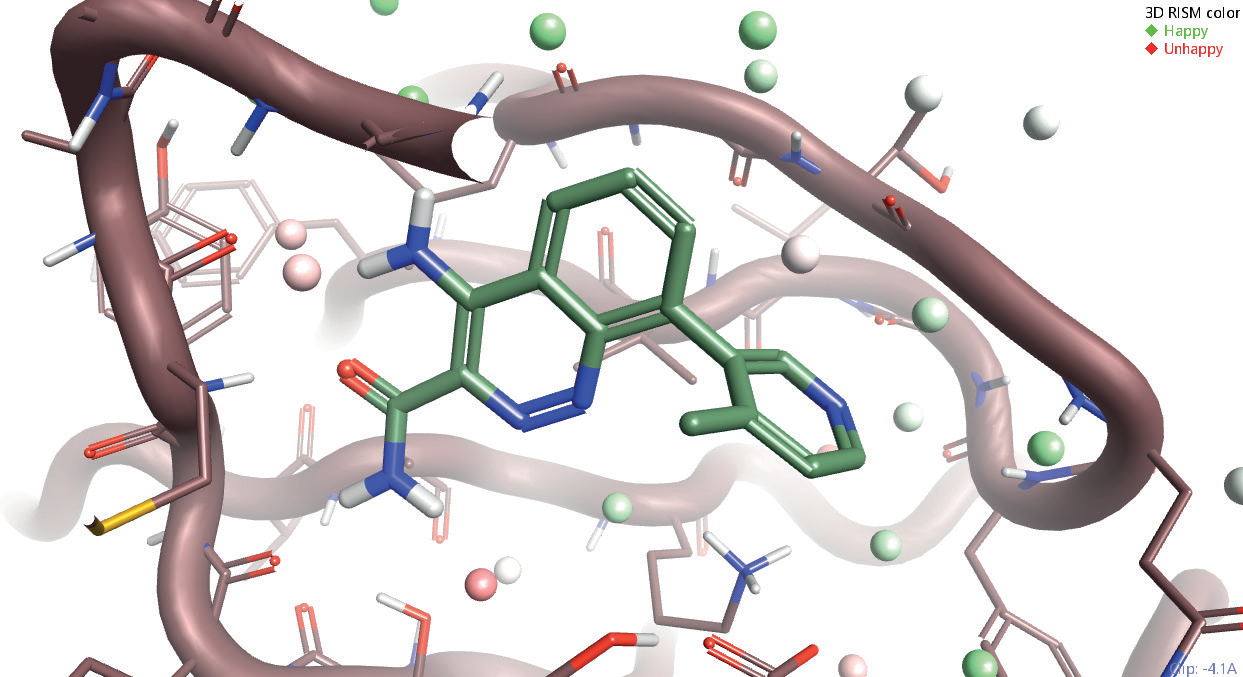
3D-RISM着色: 绿色-Happy; 红色-Unhappy
活性位点附近水分子的位置与能量学对于理解配体的结合是至关重要的。哪个水分子是紧密结合地、哪个水分子在能量上是不利的知识为结构-活性关系提供的重要信息,并可以帮您决定配体的原子应该如何摆放。Cresset的3D-RISM采用XED力场进行分析、为您提供可靠的水分子分析结果。用3D-RISM您可以:
- 理解蛋白的水能量学特征
- 即使是笔记本,也能帮你分分钟设计新的配体、理解水分子的相互作用
- 在分析的时候考虑了稳定的水分子作用,让蛋白的相互作用势更加完美
五. Lead Finder: 蛋白-配体对接计算
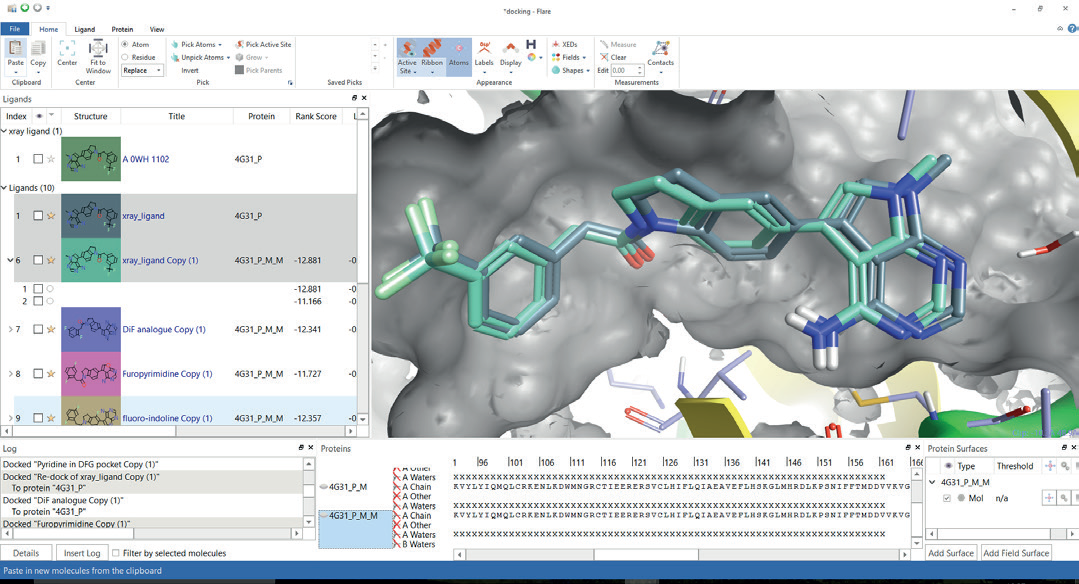
Lead Finder: 分子对接计算引擎
Lead Finder将化合物柔性地对接到静态蛋白结构里,具有非常优秀的结合模式预测性能;虚拟筛选具有高性能、高富集能力。用Lead Finder您可以:
- 通过虚拟筛选发现全新先导化合物(通过Lead Finder)
- 通过虚拟筛选设计集中库(focused libraries)
- 预测活性化合物的结合模式
- 支持共价对接计算
- 快速地拿到与活性位点匹配的新分子
跟据测试,Lead Finder比同行GOLD,GLIDE,FlexX, Surflex-Dock在同样的数据集上显著地表现出更加优秀的结合模式预测性能(re-docking精度)。
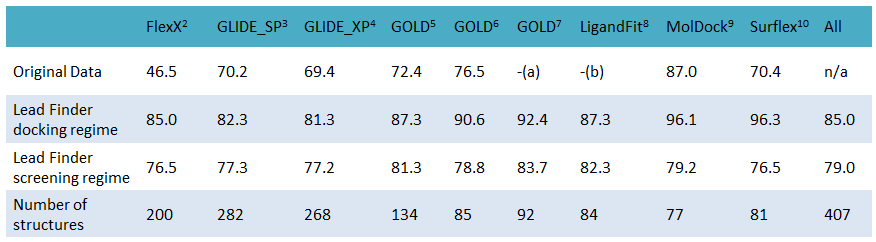
a) Data for the high-resolution subset (92 structures with resolution better than 2 Å) are not provided in original publication, only overall performance (over entire CCDC/Astex test of 224 structures) is given.
b) Original publication contains data only for 19 complexes, which is too small for comparative purposes. We extended this set by including additional 75 protein-ligand complexes which were used in original publication to benchmark LigandFit ability to detect active site cavity.
1. J. C. Cole, C. W. Murray, J.W.M. Nissink, R. D. Taylor, R. Taylor, Comparing Protein-Ligand Docking Programs Is Difficult, PROTEINS: Structure, Function, and Bioinformatics, 2005, 60, 325-332.[ Abstract ].
2. M. Rarey, B. Kramer, T. Lengauer, Multiple automatic base selection: Protein-ligand docking based on incremental construction without manual intervention J Comp Aid Mol Des, 1997, 11, 369-384. [ Abstract ].
3. R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin, D. T. Mainz, Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy, J Med Chem 2004, 47, 1739-1749.[ Abstract ].
4. R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis, P. S. Shenkin, Extra Precision Glide: Docking and Scoring Ltdorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes, J Med Chem, 2006, 49, 6177-6196.[ Abstract ].
5. G. Jones, P. Willett, R. C. Glen, A. R. Leach, R. Taylor Development and Validation of a Genetic Algorithm for Flexible Docking, J Mol Biol, 1997, 267, 727-748. [ Abstract ].
6. M. J. Hartshorn, M. L. Verdonk, G. Chessari, S. C. Brewerton, W.T..M. Mooij, P. N. Mortenson, C. W. Murray, Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance, J. Med Chem, 2007, 50, 726-741. [ Abstract ].
7. J.W.M. Nissink, C. Murray, M. Hartshorn, M. L. Verdonk, J. C. Cole, R. Taylor A NewTest Set for Validating Predictions of Protein-Ligand Interaction, PROTEINS: Structure, Function, and Genetics, 2002, 49, 457-471.[ Abstract ].
8. C. M. Venkatachalam, X. Jiang, T. Oldfield, M. Waldman LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites, J Mol Graph Model, 2003,21, 289-307. [ Abstract ].
9. R. Thomsen, M. H. Christensen, MolDock: A new technique for high-accuracy molecular docking, J Med Chem, 2006, 49, 3315-3321. [ Abstract ].
10. A. N. Jain, Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine, J Med Chem, 2003, 46, 499-511.[ Abstract ].
注意:Flare的分子对接计算引擎为Lead Finder,Flare设计用来结合模式预测与结合亲合力预测;如果需要大规模虚拟筛选,需要另外Lead Finder的license。
六. 相关主题
1.FLARE教程 | WaterSwap计算结合自由能
http://blog.molcalx.com.cn/2017/07/09/flare-tutorial-waterswap.html
2.Flare教程 | WaterSwap-如何整合公有云加速结合自由能的计算
http://blog.molcalx.com.cn/2018/02/14/cresset-broker-tutorial.html
3. FLARE案例 | WaterSwap计算BRD4抑制剂的结合自由能
http://blog.molcalx.com.cn/2018/04/15/flare-waterswap-case-study-brd4.html
七. 联系我们,获取试用: 免费!
试用下载:http://www.cresset-group.com/try-a-free-demo


