
摘要:WaterSwap是基于结构设计软件Flare的一个计算模块,专门用来计算配体-蛋白的结合自由能,它可以:1)研究配体结合的能量学特点;2)将结合能分解为每个残基的贡献以发现优选的配体相互作用模式;3) 计算一系列化合物的结合自由能(ΔG of binding)以帮助您优选配体,或者比较几种结合模式以发现最可能的结合模式。本文提供了Flare WaterSwap的计算教程。
一. 背景介绍
配体-蛋白结合不仅是生物功能也是药物化学活性的关键。有的配体直接抑制蛋白的功能,有的配体诱导蛋白构象变化进而调节关键的细胞信号通路。无论是哪一种情况,获得理想的治疗效果有赖于配体结合到靶标蛋白的结合强度。设计与靶标紧密结合而又保留安全与生物活性的配体是小分子药物发现项目的基础目标。因此,精确预测配体-蛋白结合亲合力是计算化学与计算机辅助药物设计的主要目标之一。解决这个问题最严格的方法是自由能模拟,常见的方法有:自由能微扰(free energy perturbation,FEP),热力学积分(Thermodynamic integration,TI)等等。

图1. WaterSwap用λ-坐标在两态之间”渐进渐出”
WaterSwap是基于结构药物设计软件Flare一个计算结合自由能的模块。如图1所示,WaterSwap在计算过程中构造一个反应坐标进行迭代计算:将结合到蛋白的配体“挖出”,然后用等体积的、形状的水填入。其效果是相当于将结合到蛋白的配体转移到溶剂中;同时,WaterSwap也将与配体等体积、形状的水从溶剂中转移到蛋白结合位点去。这种“挖出来”与“放进去”的路径实际就是蛋白-配体的解离、结合路径。WaterSwap采用蒙特卡洛模拟(蛋白骨架不动,侧链、配体可以自由运动)来进行构象采集,计算结合结合自由能。详细算法参见:
1. Woods, C. J., Malaisree, M., Hannongbua, S., Mulholland, A.J., “A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies”, J. Chem. Phys. 134, 054114, 2011, DOI:10.1063/1.3519057
2. Woods, C. J., Malaisree, M., Michel, J., Long, B., McIntosh-Smith, S., Mulholland, A. J., “Rapid Decomposition and Visualisation of Protein-Ligand Binding Free Energies by Residue and by Water”, Faraday Discussions 169: Molecular Simulation and Visualisation, 2014, DOI:10.1039/C3FD00125C
WaterSwap可以用来:
- 精确计算配体-受体结合自由能
- 预测结合水与可替换水分子位置,用来指导结构改造,提高化合物的结合亲和力与选择性
- 能量拆解,可视化分析帮助理解残基对结合的贡献
二. 操作步骤
1. 结构准备
WaterSwap计算的精度取决于复合物结构模型,如果复合物结构模型错误,计算也就不值一提。
在计算之前需要准备蛋白与配体结构。其中蛋白结构可以从蛋白-配体共晶复合物结构(HOLO结构)开始准备,也可以从一个没有结合配体的蛋白结构(APO结构)开始准备。Flare提供了Protein Preparation工具,可以顺利的完成这个准备过程。而配体则必须是一个正确的pose,可以从复合物结构里提取而得,也可以是docking的一个pose或分子叠合的一个pose。本例假设从PDB上下载的复合物结构4ZLZ开始。
如何准备蛋白结构与对接,请参见Flare教程|分子对接-结合模式预测。
2. WaterSwap计算
准备好配体与受体结构之后,就可以开始用WaterSwap计算结合自由能。WaterSwap在Flare菜单Ligand>WaterSwap下,见图2。
图 2. WaterSwap菜单
点击WaterSwap菜单后,弹出WaterSwap计算对话框,见图3。根据要计算的蛋白与配体对进行设定。

图3. WaterSwap计算对话框
在Chain选项框里,Other通常为金属、Cofactor或无关的辅剂(比如共晶实验用到的糖等)。假设不需要在计算中考虑这些分子,则去掉该选项。假如Cofactor需要考虑、金属需要考虑,糖不需要包括在计算里,请在上一步结构准备的时候,在Protein的窗口里,删除糖就可以了。
WaterSwap建议采用至少1000个迭代之后的收敛,因此将Interation设置为1000,并将Stop on Converge选项关闭:以便确保至少是1000个迭代之后收敛。如果到达1000个迭代之后还没有收敛,可以在WaterSwap界面点击Continue继续计算直到收敛。
点击WaterSwap计算对话框的Show Option,有更多的参数可供选择(图4)。WaterSwap的电荷模型可选Gasteiger或AM1-BCC;其中OpenMM部分(分子动力学模拟部分)计算平台可选CPU或GPU(推荐选CPU,因为比例很低),设定好选项之后,点击Start开始计算。
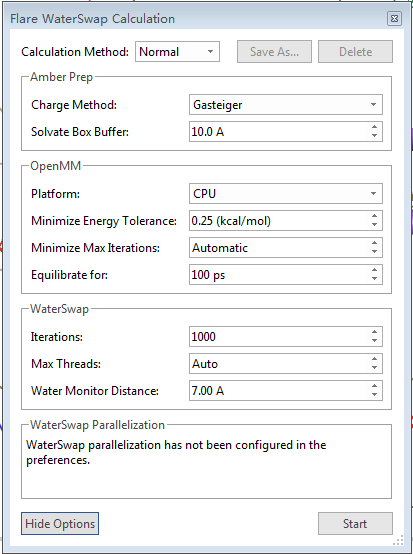
图4. WaterSwap计算选项(OpenMM部分虽然有GPU可选,但是只用来做约束的分子动力学模拟,占用机时非常少)
WaterSwap的电荷模型可选Gasteiger或AM1-BCC;OpenMM分子动力学模拟部分计算平台可选CPU或GPU。设定好选项之后,点击Start开始计算,并弹出计算的进度与状态(图5)。
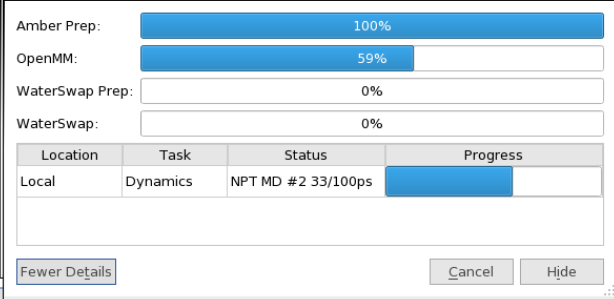
图5. WaterSwap计算进度
注意:WaterSwap需要很大量的计算资源,以PDB代码为4ZLZ的配体-蛋白复合物晶体结构为例,在一个Linux工作站(CPU:Intel XEON E5-2683 V3 2.0GHz,系统内存:128GB)上,用28核心计算,需要20个小时。普通的笔记本或办公电脑采用(比如i7-4700)需要几天或更长时间,因此不要在笔记本上计算!
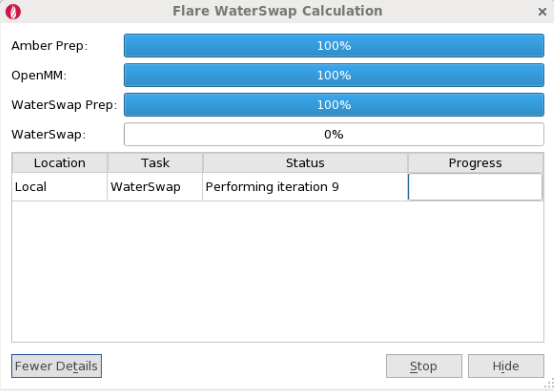
图6. WaterSwap迭代
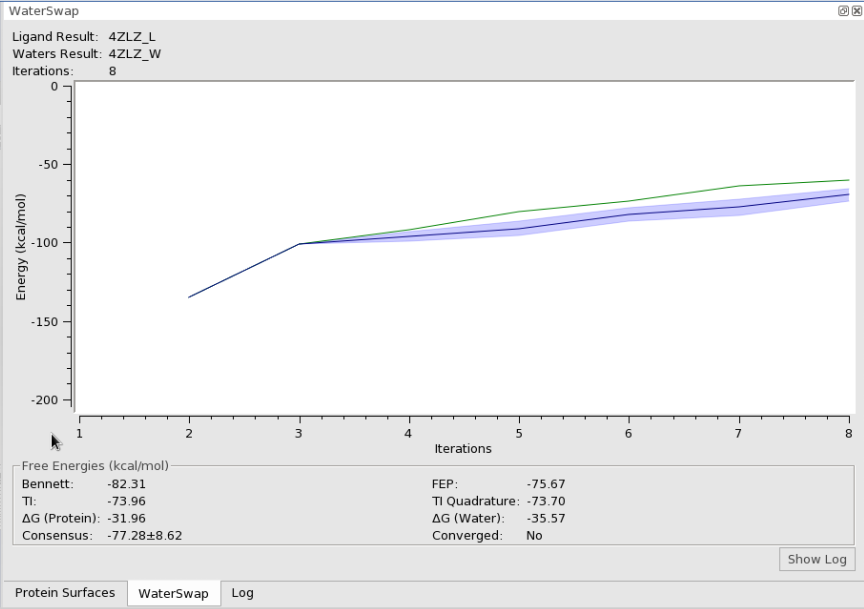
图7. WaterSwap计算的结合自由能
我们也会从Flare的进度窗口上看到WaterSwap不断在进行迭代计算(图6),每个迭代都给出4中不同的算法的自由能(图7):
- Benneet:用Bennett’s Acceptance Ratio (BAR)计算的结合自由能
- TI: 用Thermodynamic Intergration计算的结合自用能
- FEP:用Free Energy Peturbation计算的结合自由能
- TI Quadrature:用Thermodynamic Intergration with numerical quadrature计算的结合自由能
- ΔG(Protein):Total free energy of binding due to the protein box
- ΔG(Water): Total free energy of binding due to the water box
- Consensus: Consensus free energy of binding,是上述四种结合自由能的加权平均。
- Converged: 根据四种自由能算法计算ΔG相似性程度的一种收敛指标。注意,这并非硬标准,应该去检查轨迹以判断是否收敛。
图7是WaterSwap迭代计算的监控结果,一直迭代750次后达到收敛标准。
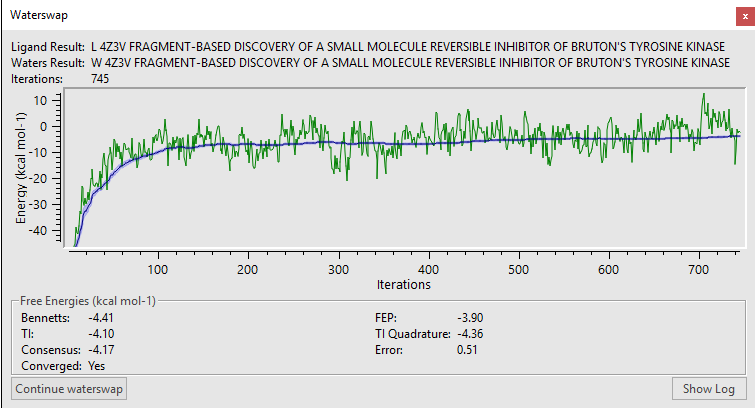
图7. WaterSwap计算的收敛与结束。当结合自由能值波动(最大与最小值差)小于1.5kcal/mol时达到收敛,WaterSwap计算结束。本次计算共迭代了758次才达到收敛标准。
三. WaterSwap可视化结果分析
WaterSwap计算完毕,在蛋白表单会新添加两个蛋白,其命名代表着计算性质:如果蛋白名字有”_L”结尾,代表该蛋白是最后一帧动态快照,含有配体,其颜色按WaterSwap能量系数着色;如果名字是”_W”结尾,代表该蛋白含水盒子,其颜色也是按WaterSwap能量系数着色。
配体、水周围的蛋白氨基酸残基按WaterSwap的能量用绿色到红色渐变标度(图8):
- 绿色越浓的氨基酸残基越倾向于与配体发生结合;
- 白色氨基酸残基对结合没有贡献或其没有WaterSwap系数;
- 红色越浓的氨基酸残基越偏向于水发生相互作用。
配体-蛋白(蛋白_L)可视化分析可以告诉我们配体的结合自由能贡献来源:绿色氨基酸残基对配体贡献了大部分的结合自由能;红色的氨基酸残基发生去溶剂化而对配体的结合起不利作用。
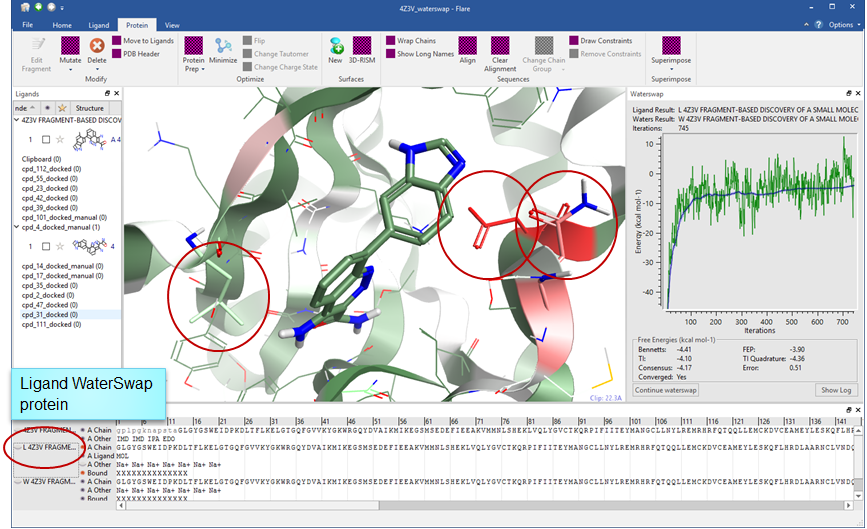
图8. WaterSwap可视化分析残基对结合的贡献
水-蛋白(蛋白_W)可视化分析可以告诉我们:在蛋白的活性位点里,哪个地方是水优选的结合位置。水分成两个链:Swap Water与Bound Water。Swap Water是与配体发生互换的水,与配体一起观察,可以帮助在配体设计时发现活性位点里配体应该发生引入亲水基团的地方。Bound Water是那些在结合位点里出现在配体周围的水或水盒子里的水,它们的颜色按能量着色。
四. 注意事项
- WaterSwap是计算绝对结合自由能,在计算中忽略了可极化性、结合位点的蛋白部分骨架不动,所以WaterSwap计算的结合自由能是高估的,不能与实验自由能直接进行比较。但是可以计算一些列化合物的结合自能,将之与实验值进行拟合。
- WaterSwap计算量很大,为保证在合理的时间内得到结果,建议采用单机多核计算。在我们的测试里,单节点含28核心可以在合理的时间内给出计算结果。
- 在进行计算时,WaterSwap默认会将全部CPU用于计算。
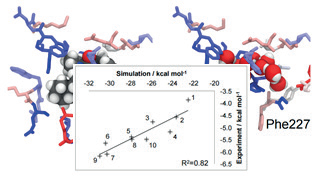
图9. WaterSwap计算系列化合物的结合自由能,并与实验值拟合
WaterSwap计算阶段,虽然进行蒙特卡罗模拟(Monte Carlo simulation),但是熵的计算是不完全的,主要是因为:1)蛋白骨架是被约束不动的,仅结合位点部分的侧链可以发生运动。大的蛋白构象变化不会被采集到,因此蛋白的构象熵计算不是完全的;2)在蛋白结合位点里配体虽然可自由移动,但也不会发生取向截然不同的情况,WaterSwap的精度取决于复合物模型的精度。如果复合物模型一开始就是错的,那WaterSwap的结合也不值一提。
WaterSwap初始是用OpenMM计算,可以用GPU加速,但是这部分实际上耗时较少。WaterSwap计算是最耗时阶段,仅支持CPU计算。
四. 文献
方法学文献
Woods, C. J., Malaisree, M., Hannongbua, S., Mulholland, A.J., “A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies”, J. Chem. Phys. 134, 054114, 2011, DOI:10.1063/1.3519057
Woods, C. J., Malaisree, M., Michel, J., Long, B., McIntosh-Smith, S., Mulholland, A. J., “Rapid Decomposition and Visualisation of Protein-Ligand Binding Free Energies by Residue and by Water”, Faraday Discussions 169: Molecular Simulation and Visualisation, 2014, DOI:10.1039/C3FD00125C
应用文献
Woods, C. J.; Malaisree, M.; Long, B.; McIntosh-Smith, S.; Mulholland, A. J. Computational assay of H7N9 influenza neuraminidase reveals R292K mutation reduces drug binding affinity. Scientific reports 2013, 3, 3561. DOI:10.1038/srep03561
五. 相关教程
1. 《Flare教程 | WaterSwap-如何整合公有云加速结合自由能的计算》
2. 《FLARE案例 | WaterSwap计算BRD4抑制剂的结合自由能》
六. 硬件配置与收益
1. 硬件配置
8个节点,每个节点关键指标见表1。
总价格:30万
表1. 每个节点的关键指标
| 名称 | 规格 | 数量 |
| CPU | Intel Xeon E5-2686 V4 SR2K8 2.30GHz 18-Core Broadwell Processor 45MB L3 Cache CPU | 2 颗 |
| 内存 | 64GB | – |
2. 预期收益
- 每个节点并发2个18核心作业
- 8个节点并发计算1个作业
则8个节点共并发16个作业,每个作业2-3天完成,因此预期平均5-8个化合物/天。
预期计算速度为:3-5小时/化合物。
七. 试用与下载
试用下载:http://www.cresset-group.com/try-a-free-demo


