
摘要:本文主要提供pyflare的python extension与rdkit等的常见问题。包括如何(1)根据蛋白的UNIPROT批量下载PDB结构,(2)根据PDB ID下载PDB结构;(3)如何用RDKit计算指纹图谱、相似性搜索、子结构搜索等等。
本文专门回答Flare的python extension、RDKit相关问题。
1. 如何根据蛋白的UNIPROT获取号获得PDB数据库的PDB code
问题:我对EGFR激酶感兴趣,我想将全部的EGFR激酶下载下来,然后用FLARE准备,再用AutoT&T进行从头设计分子。那么我该如何获取EGFR激酶的PDB ID号呢?
答:假设你对人源的EGFR激酶感兴趣,它对应的UNIPROT AC号为P00533,我们可以用P00533为关键词获得全部的PDB结构。代码如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 | import requests import time uniprot_ids = ['P00533'] url = 'https://www.uniprot.org/uniprot/' protein_to_pdb = {} for protein in uniprot_ids: params = { 'format': 'tab', 'query': 'ID:{}'.format(protein), 'columns': 'id,database(PDB)' } contact = "" # Please set your email address here. headers = {'User-Agent': 'Python {}'.format(contact)} r = requests.get(url, params=params, headers=headers) protein_to_pdb[protein] = str(r.text).splitlines()[-1].split('\t')[-1].split(';') protein_to_pdb[protein].pop(-1) time.sleep(1) # be respectful and don't overwhelm the server with requests print(protein_to_pdb) |
结果如下:
1 | {'P00533': ['1DNQ', '1DNR', '1IVO', '1M14', '1M17', '1MOX', '1NQL', '1XKK', '1YY9', '1Z9I', '2EB2', '2EB3', '2EXP', '2EXQ', '2GS2', '2GS6', '2GS7', '2ITN', '2ITO', '2ITP', '2ITQ', '2ITT', '2ITU', '2ITV', '2ITW', '2ITX', '2ITY', '2ITZ', '2J5E', '2J5F', '2J6M', '2JIT', '2JIU', '2JIV', '2KS1', '2M0B', '2M20', '2N5S', '2RF9', '2RFD', '2RFE', '2RGP', '3B2U', '3B2V', '3BEL', '3BUO', '3C09', '3G5V', '3G5Y', '3GOP', '3GT8', '3IKA', '3LZB', '3NJP', '3OB2', '3OP0', '3P0Y', '3PFV', '3POZ', '3QWQ', '3UG1', '3UG2', '3VJN', '3VJO', '3VRP', '3VRR', '3W2O', '3W2P', '3W2Q', '3W2R', '3W2S', '3W32', '3W33', '4G5J', '4G5P', '4HJO', '4I1Z', '4I20', '4I21', '4I22', '4I23', '4I24', '4JQ7', '4JQ8', '4JR3', '4JRV', '4KRL', '4KRM', '4KRO', '4KRP', '4LI5', '4LL0', '4LQM', '4LRM', '4R3P', '4R3R', '4R5S', '4RIW', '4RIX', '4RIY', '4RJ4', '4RJ5', '4RJ6', '4RJ7', '4RJ8', '4TKS', '4UIP', '4UV7', '4WD5', '4WKQ', '4WRG', '4ZAU', '4ZJV', '4ZSE', '5C8K', '5C8M', '5C8N', '5CAL', '5CAN', '5CAO', '5CAP', '5CAQ', '5CAS', '5CAU', '5CAV', '5CNN', '5CNO', '5CZH', '5CZI', '5D41', '5EDP', '5EDQ', '5EDR', '5EM5', '5EM6', '5EM7', '5EM8', '5FED', '5FEE', '5FEQ', '5GMP', '5GNK', '5GTY', '5GTZ', '5HCX', '5HCY', '5HCZ', '5HG5', '5HG7', '5HG8', '5HG9', '5HIB', '5HIC', '5J9Y', '5J9Z', '5JEB', '5LV6', '5SX4', '5SX5', '5U8L', '5UG8', '5UG9', '5UGA', '5UGB', '5UGC', '5UWD', '5WB7', '5WB8', '5X26', '5X27', '5X28', '5X2A', '5X2C', '5X2F', '5X2K', '5XDK', '5XDL', '5XGM', '5XGN', '5XWD', '5Y25', '5Y9T', '5YU9', '5ZWJ', '6ARU', '6B3S', '6D8E']} |

2. 如何根据PDB ID号下载PDB结构
答:用个下载的helper吧:
1 2 3 4 5 | def download_from_pdb(project, code): url = 'http://files.rcsb.org/view/{0:s}.pdb'.format(code) response = urllib.request.urlopen(url) text = response.read().decode('utf-8') project.proteins.extend(flare.read_string(text, 'pdb')) |
假设你的Flare项目名称为p,将PDB code为1eve的PDB结构下载读入到Flare的视窗:
1 | download_from_pdb(p, '1eve') |
如果想下载PDB到硬盘而不用flare里,那就更简单:
1 2 3 4 5 6 7 8 | def download_from_pdb(code): from tqdm import tqdm import requests url = 'http://files.rcsb.org/view/{0:s}.pdb'.format(code) response = requests.get(url, stream=True) with open(code+'.pdb', "wb") as handle: for data in tqdm(response.iter_content()): handle.write(data) |
3. 计算一个SMILES格式数据库的MORGAN_2 512BIT长度指纹图谱
答:下面的python这个代码可以演示该功能。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 | import sys,string,argparse from optparse import OptionParser import os, gzip from rdkit import Chem from rdkit.Chem import AllChem import numpy as np parser = argparse.ArgumentParser(description='Compute Morgan2 512BIT Fingerprint') parser.add_argument('dbase',metavar='molecular database in SMILES') args = parser.parse_args() dbase = args.dbase file = open(dbase,'r') lines = file.readlines() file.close() n = len(lines) for i in range(0,n): smi = lines[i].split()[0].replace('$','') id = lines[i].split()[1].replace('$','') info = {} mol = Chem.MolFromSmiles(smi) fp = AllChem.GetMorganFingerprintAsBitVect(mol, useChirality=True, radius=2, nBits = 512, bitInfo=info) vector = np.array(fp) print(smi,id,vector) |
把上面代码复制、保存为smi2morgan512.py就可以用了。下面是一个算例: 输入一个SMILES文件(query.smi, 包含SMILES与ID), 输出SMILES、ID与512BIT指纹图谱。
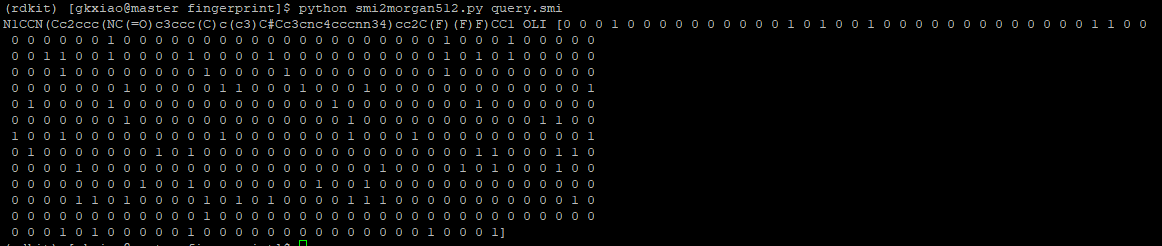
4. 如何用RDKit的Morgan 2指纹图谱进行基于2D相似性的虚拟筛选
答:假设有一个查询结构,一个数据库,计算查询结构与数据库化合物的之间的Morgan 2相似性,根据相似性进行过滤即可。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 | import sys,string,argparse from optparse import OptionParser import os, gzip from rdkit import Chem from rdkit.Chem import AllChem from rdkit import DataStructs import numpy as np parser = argparse.ArgumentParser(description='Compute Morgan2 Similarity between query and database moelecules') parser.add_argument('query',metavar='<query moleclue>') parser.add_argument('dbase',metavar='<database in SMILES>') args = parser.parse_args() query = args.query dbase = args.dbase query = open(args.query).readline().split()[0].replace('$','') qmol = Chem.MolFromSmiles(query) qfp = AllChem.GetMorganFingerprintAsBitVect(qmol, useChirality=True, radius=2, nBits = 1024) file = open(dbase,'r') lines = file.readlines() file.close() n = len(lines) for i in range(0,n): try: smi = lines[i].split()[0].replace('$','') id = lines[i].split()[1].replace('$','') mol = Chem.MolFromSmiles(smi) fp = AllChem.GetMorganFingerprintAsBitVect(mol, useChirality=True, radius=2, nBits = 1024) print(smi,id,DataStructs.FingerprintSimilarity(qfp,fp)) except: pass |
使用方法:将上述文件保存为similarity.py, 键入如下命令:
1 | pyflare similarity.py --help |
假设查询结构(参比分子)为query.smi,数据库为DDR1.smi,则键入如下命令:
1 | pyflare ./similarity.py query.smi DDR1.smi |
上述命令会计算DDR1.smi里每一个化合物与query.smi化合物的相似性值,给出类似如下结果:其中第1列为SMILES代码,第2列ID号,第3列为相似性值
1 2 3 4 5 6 7 8 9 10 11 12 | C#Cc1cccc(NC(=O)c2cccc(Cl)c2)c1 ZINC000023055325 0.2 CSc1ccc(C(=O)Nc2cccc(C(F)(F)F)c2)cc1 ZINC000023120080 0.25 NC(=O)c1ccc(C(=O)Nc2cccc(C(F)(F)F)c2)cc1 ZINC000023120087 0.24705882352941178 COCc1ccc(C(=O)Nc2cccc(C(F)(F)F)c2)cc1 ZINC000023120089 0.25555555555555554 CC(C)OCc1ccc(C(=O)Nc2cccc(C(F)(F)F)c2)cc1 ZINC000023120090 0.25 Cc1nc(-c2cccc(NC(=O)c3ccc(Cl)cc3)c2)cs1 ZINC000023135735 0.20833333333333334 CC(=O)Nc1ccc(C(=O)Nc2cccc(-c3csc(C)n3)c2)cc1 ZINC000023135776 0.22105263157894736 CCc1ccc(C(=O)Nc2cccc(-c3csc(C)n3)c2)cc1 ZINC000023135876 0.21875 CCCOc1ccc(C(=O)Nc2cccnc2)cc1OCCC ZINC000023136185 0.2808988764044944 Cc1ccc(C(=O)NCCNC(=O)CN2CCN(Cc3ccccc3)CC2)cc1F ZINC000023140343 0.30434782608695654 CSc1ccc(C(=O)Nc2ccnn2Cc2ccccc2)cc1[N+](=O)[O-] ZINC000023141397 0.26262626262626265 Cc1ccc(C(=O)Nc2ccnn2Cc2ccccc2)cc1[N+](=O)[O-] ZINC000023141612 0.3010752688172043 |
5. 如何进行子结构搜索
答:SMARTS语言可以用来定义子结构,并进行数据库搜索。下面的代码可以实现该过程。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 | import sys,string,argparse from optparse import OptionParser import os, gzip from rdkit import Chem from rdkit.Chem import AllChem from rdkit import DataStructs import numpy as np parser = argparse.ArgumentParser(description='Substructure Search') parser.add_argument('query',metavar='<SMARTS query>') parser.add_argument('dbase',metavar='<molecular database in SMILES>') args = parser.parse_args() query = args.query dbase = args.dbase query = Chem.MolFromSmarts(query) file = open(dbase,'r') lines = file.readlines() file.close() n = len(lines) for i in range(0,n): try: smi = lines[i].split()[0].replace('$','') id = lines[i].split()[1].replace('$','') mol = Chem.MolFromSmiles(smi) if mol.HasSubstructMatch(query): print(smi,id) except: pass |
用法:将上述代码保存为subsearch.py,键入如下命令:
1 | python subsearch.py --help |
具体例子:搜索DDR1数据库中的具有苯环的化合物结构。
1 | python ./subsearch.py c1ccccc1 DDR1.smi |
部分结果如下:
1 2 3 4 5 6 7 8 9 10 11 | CCOc1cc2ncnc(Nc3cccc(C#N)c3)c2cc1OCC ZINC000026189586 Cc1ccc(C(=O)NCCc2nccn2Cc2ccccc2)cc1[N+](=O)[O-] ZINC000026214908 Cc1cc(C(=O)NCCc2nccn2Cc2ccccc2)ccc1[N+](=O)[O-] ZINC000026215912 COc1ccc(C(=O)Nc2cccc(CNCCN3CCN(Cc4ccccc4)CC3)c2)cc1 ZINC000026312311 O=C(Nc1cccc(C#Cc2ccccn2)c1)c1ccc(-n2ccnc2)nc1 ZINC000026314197 C[C@@H](NCc1cccc(NC(=O)c2ccc(Cl)cc2)c1)c1cccnc1 ZINC000026317864 C[C@H](NCc1cccc(NC(=O)c2ccc(Cl)cc2)c1)c1cccnc1 ZINC000026317867 COc1cccc(NC(=O)c2cc(C)no2)c1 ZINC000026359780 COc1cccc(C(=O)NCCNC(=O)c2ccc(-n3nccc3C)cc2)c1 ZINC000026375600 COc1cccc(C(=O)NCCNC(=O)c2ccc(C#Cc3ccccc3)cc2)c1 ZINC000026376732 COc1cccc(C(=O)NCCNC(=O)c2ccc(NC(=O)c3ccccc3)cc2)c1 ZINC000026377855 |
6. shell逐行处理SMILES格式的问题
用echo组合read逐行处理SMILES文件会遇到转义问题, 错误使用会出现化合物立体化学信息丧失问题。比如下面的demo.smi含两个顺反异构体化合物:
1 2 | FC(F)(/C([H])=C(C(F)(F)F)\[H])F ZC4F6 FC(F)(/C([H])=C([H])\C(F)(F)F)F EC4F6 |
下面的read与echo命令会让“\”发生转义:
1 2 3 4 5 6 | [gkxiao@master tangnian]$ cat demo.smi |while read line > do > echo $line > done FC(F)(/C([H])=C(C(F)(F)F)[H])F ZC4F6 FC(F)(/C([H])=C([H])C(F)(F)F)F EC4F6 |
注意:顺反异构体都变成同一个化合物了!这主要是read引起的,可以加-r参数来避免这个问题:
1 2 3 4 5 6 | [gkxiao@master tangnian]$ cat demo.smi |while read -r line > do > echo $line > done FC(F)(/C([H])=C(C(F)(F)F)\[H])F ZC4F6 FC(F)(/C([H])=C([H])\C(F)(F)F)F EC4F6 |
7. RDKit读取SDF时氢被删除的问题
问题:RDKit用SDMolSupplier读入SDF文件时氢被删除,虽然可以用AddHs()补上,但是这个氢不是原先的氢。
解决方法1: 将removeHs关闭
1 2 | from rdkit import Chem suppl = Chem.SDMolSupplier('test.sdf', removeHs=False) |
解决方法2:文本转结构
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
from __future__ import print_function
from rdkit import Chem
#读入sdf文件,本例子里只有一个化合物苯
suppl = Chem.SDMolSupplier('/upload/gkxiao/test.sdf')
#suppl得到的mol,氢被删除
mol1 = suppl[0]
print(Chem.MolToMolBlock(mol1))
#结果如下,氢不见了
bene
RDKit 2D
6 6 0 0 0 0 0 0 0 0999 V2000
-0.8660 -0.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7321 -0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7321 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.8660 1.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 6 2 0
1 2 1 0
2 3 2 0
3 4 1 0
4 5 2 0
5 6 1 0
M END
#读取第一个化合物为一个文本
block = suppl.GetItemText(0)
#将文本转为分子
mol2 = Chem.MolFromMolBlock(block,removeHs=False,sanitize = True)
#这样氢还在
print(Chem.MolToMolBlock(mol2))
bene
RDKit 2D
12 12 0 0 0 0 0 0 0 0999 V2000
-0.8660 -0.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7321 -0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.7321 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.8660 1.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.8660 -1.5320 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.6258 -0.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.6258 1.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.8660 2.5320 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.8937 1.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.8937 -0.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1 6 2 0
1 2 1 0
1 7 1 0
2 3 2 0
2 8 1 0
3 4 1 0
3 9 1 0
4 5 2 0
4 10 1 0
5 6 1 0
5 11 1 0
6 12 1 0
M END
8. 用rkdit从SMILES生成3D结构
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 | from __future__ import print_function from rdkit import Chem #读入sdf文件,本例子里只有一个化合物苯 suppl = Chem.SDMolSupplier('/upload/gkxiao/test.sdf') #suppl得到的mol,氢被删除 mol1 = suppl[0] print(Chem.MolToMolBlock(mol1)) #结果如下,氢不见了 bene RDKit 2D 6 6 0 0 0 0 0 0 0 0999 V2000 -0.8660 -0.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.7321 -0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.7321 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.8660 1.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.0000 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1 6 2 0 1 2 1 0 2 3 2 0 3 4 1 0 4 5 2 0 5 6 1 0 M END #读取第一个化合物为一个文本 block = suppl.GetItemText(0) #将文本转为分子 mol2 = Chem.MolFromMolBlock(block,removeHs=False,sanitize = True) #这样氢还在 print(Chem.MolToMolBlock(mol2)) bene RDKit 2D 12 12 0 0 0 0 0 0 0 0999 V2000 -0.8660 -0.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.7321 -0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.7321 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.8660 1.5000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.0000 1.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.8660 -1.5320 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 -2.6258 -0.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 -2.6258 1.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 -0.8660 2.5320 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 0.8937 1.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 0.8937 -0.5160 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 1 6 2 0 1 2 1 0 1 7 1 0 2 3 2 0 2 8 1 0 3 4 1 0 3 9 1 0 4 5 2 0 4 10 1 0 5 6 1 0 5 11 1 0 6 12 1 0 M END |
从SMILES生成3D结构的演示,原文请参考:http://www.rdkit.org/docs/GettingStartedInPython.html
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 | from rdkit import Chem from rdkit.Chem import AllChem >>> smi = 'C1CCC1' >>> m1 = Chem.MolFromSmiles(smi) >>> m1.SetProp("_Name","cyclobutane") >>> print(Chem.MolToMolBlock(m1)) cyclobutane RDKit 2D 4 4 0 0 0 0 0 0 0 0999 V2000 1.0607 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.0000 -1.0607 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.0607 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 0.0000 1.0607 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1 2 1 0 2 3 1 0 3 4 1 0 4 1 1 0 M END #加氢,生成3D结构 >>> m2 = Chem.AddHs(m1) >>> AllChem.EmbedMolecule(m2) >>>print(Chem.MolToMolBlock(m2)) cyclobutane RDKit 3D 12 12 0 0 0 0 0 0 0 0999 V2000 0.4280 0.9580 -0.0542 C 0 0 0 0 0 0 0 0 0 0 0 0 0.9494 -0.4476 0.0775 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.4453 -0.9477 -0.1840 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.9600 0.4286 0.2195 C 0 0 0 0 0 0 0 0 0 0 0 0 0.8336 1.6583 0.6806 H 0 0 0 0 0 0 0 0 0 0 0 0 0.5192 1.3314 -1.1071 H 0 0 0 0 0 0 0 0 0 0 0 0 1.6987 -0.6701 -0.7153 H 0 0 0 0 0 0 0 0 0 0 0 0 1.3564 -0.7346 1.0500 H 0 0 0 0 0 0 0 0 0 0 0 0 -0.7635 -1.7281 0.5135 H 0 0 0 0 0 0 0 0 0 0 0 0 -0.5819 -1.1411 -1.2610 H 0 0 0 0 0 0 0 0 0 0 0 0 -1.7380 0.7914 -0.4921 H 0 0 0 0 0 0 0 0 0 0 0 0 -1.2964 0.5016 1.2727 H 0 0 0 0 0 0 0 0 0 0 0 0 1 2 1 0 2 3 1 0 3 4 1 0 4 1 1 0 1 5 1 0 1 6 1 0 2 7 1 0 2 8 1 0 3 9 1 0 3 10 1 0 4 11 1 0 4 12 1 0 M END |
9. 从其它格式生成高斯输入文件
问:如何从SMILES结构批量生成Gaussian输入文件,包括3D结构生成、结构优化、生成输入文件。
答:假设输入文件为dbase.smi,含有多个化合物,又假设拟采用Gaussian在apfd/6-311+g(2d,p)理论水平进行优化与频率计算,可以用OpenBabel进行格式转化:
1 | obabel -ismi dbase.smi -ogjf -O dbase_.com --gen3d -m --minimize --ff MMFF94 -xk '#p opt freq apfd/6-311+g(2d,p)' |
其中-xk为Gaussian的route行关键字;dbase_是生成文件的文件名前缀。
该命令生成了一系列Gaussian输入文件:dbase_1.com, dbase_2.com, dbase_3.com, … , dbase_n.com。典型的内容如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | #p opt freq apfd/6-311+g(2d,p) demo 0 1 C 1.07095 0.01052 -0.05662 C 2.40659 0.01052 -0.05662 H 0.51134 -0.90289 0.11902 H 0.51134 0.92393 -0.23226 H 2.96621 -0.90289 0.11902 H 2.96621 0.92393 -0.23226 !前面空白行 |
如果不需要进行3D结构生成与结构优化,可以直接生成Gaussian输入文件:
1 | obabel -imol JMS-001.mol -ogjf -O JMS-001.com -xk '#p opt freq apfd/6-311+g(2d,p)' |
10. RDKit进行立体化学枚举
有时化合物SMILES代码存在缺失立体化学信息的情况,比如双键的顺反与手性中心的类型不详等等。此时需要对化学结构进行立体化学的枚举,其中一个策略是:仅对立体化学信息不明的部分进行枚举,其余部分保留原有的立体化学构型。用RDKit实现代码如下:
1 2 3 4 5 6 | from __future__ import print_function from rdkit import Chem from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers, StereoEnumerationOptions mol = Chem.MolFromSmiles('FC=CC(F)Cl') opts = StereoEnumerationOptions(onlyUnassigned=True,unique=True,maxIsomers=0) isomers = tuple(EnumerateStereoisomers(mol, options=opts)) |
其中onlyUnassigned=True表示立体化学已经指定的部分保留不变,仅枚举其余部分。其中maxIsomers=0是输出所有的异构体;如果需要随机输出一个结构,则用maxIsomers=1。
输出枚举后的SMILES
1 2 3 | for x in isomers: smi = Chem.MolToSmiles(x, isomericSmiles=True) print(smi) |
结果如下:
1 2 3 4 | F/C=C/[C@H](F)Cl F/C=C/[C@@H](F)Cl F/C=C\[C@H](F)Cl F/C=C\[C@@H](F)Cl |
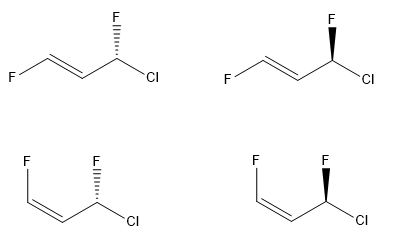
枚举生成的四个化合物结构
11. 如何用openbabel对smiles数据库去重
问题:如何检查化合物数据是否有重复的化合物结构,并去掉其中重复的化合物。
答:假设有个化合物数据库dbase.smi,可以用openbabel来去重复,命令如下:
1 | obabel -ismi dbase.smi -osmi -O dbase_nodup.smi --unique |
在屏幕上有标准输出重复的分子,并将无重复的结构保存为dbase_nodup.smi。屏幕输出的提示如下:
1 2 3 4 5 6 | Removed QBW-121 - a duplicate of YFQ-156 (#141) Removed QBW-122 - a duplicate of YFQ-005 (#142) Removed QBW-123 - a duplicate of TKM-487 (#143) Removed QBW-124 - a duplicate of YFQ-030 (#144) Removed QBW-125 - a duplicate of TDY-200602 (#145) Removed QBW-126 - a duplicate of TKM-413 (#146) |
这说明化合物QBW-121与化合物YFQ-156重复而被删除,将不会保存在输出文件dbase_nodup.smi里。
如果有数据库a.smi、b.smi, 需要得到合并后不重复的化合物。则可以先合并为dbase.smi, 再去重。合并方法如下:
1 | cat a.smi b.smi >> dbase.smi |
12. 从Flare导出3D-RISM预测的水分子位置及其ΔG
这个需要用Flare/Python解释器里,执行下面python代码,将3D-RISM的值转为温度因子。具体操作为:
(1)点击Flare | Python | Python interpretrr, 打开python解释器;
(2)将下面的代码复制、黏贴到打开python解释器里;
1 2 3 4 5 6 7 | from cresset import flare project = flare.main_window().project for prot in flare.main_window().selected_proteins: for atom in prot.atoms: dG = prot.rism_result.atom_delta_g(atom) if dG is not None: atom.tf = dG |
(3)选择中需要导出的蛋白,点击python解释器下方的run运行python代码即可。然后,再选中蛋白,export为PDB文件。
13. 判断化合物是否仅包含特定元素
问题:如何将仅由C,H,O,N元素组成的化合物过滤出来?
答:这等同于判断一个化合物是否包含一个指定元素列表之外的元素。或者给出一个允许的元素列表,判断一个化合物是否包含该列表之外的元素。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 | from rdkit import Chem m = Chem.MolFromSmiles('C1CCCCN1') def IsOnlyElement(mol): """ 如果化合物仅包含允许元素,则返回1;否则返回0 """ elements=[] for atom in mol.GetAtoms(): elements.append(atom.GetSymbol()) onlyElements=['H','C','O','N','S','P','F','Cl','Br','I'] elements = list(set(elements)) excludedElement = list(set(elements).difference(set(onlyElements))) if excludedElement : return 0 else: return 1 IsOnlyElement(m) |
1 | 1 |
14. FPSim2的相似性筛选
FPSim2源代码:https://github.com/chembl/FPSim2
创建数据库:
1 2 | from FPSim2.io import create_db_file create_db_file('chembl.smi', 'chembl.h5', 'Morgan', {'radius': 2, 'nBits': 2048}) |
相似性搜索:
1 2 3 4 5 6 7 | from FPSim2 import FPSim2Engine fp_filename = 'chembl_27.h5' fpe = FPSim2Engine(fp_filename) query = 'CC(=O)Oc1ccccc1C(=O)O' results = fpe.similarity(query, 0.7, n_workers=1) |
15. 准备好的SDF文件转化为virtualflow样式
假设数据库已经准备为sdf格式,化合物的title为id+num(需为独一无二,其中id一般为索引号,id是准备过程中扩增出来的,比如互变异构体枚举,质子化枚举)。那么下面脚本将该sdf文件按virtual-flow的tranches方式保存为单个的pdbqt格式。进一步经过打包,可用于虚拟筛选。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 | from __future__ import print_function from rdkit import Chem from rdkit.Chem import Descriptors import sys import os ifile = sys.argv[1] suppl = Chem.SDMolSupplier(ifile,removeHs=False) ofile = sys.argv[2] w = Chem.SDWriter(ofile) for id in range(len(suppl)): mol = suppl[id] if mol is None:continue mw = Descriptors.MolWt(mol) tpsa = Descriptors.TPSA(mol) logp = Descriptors.MolLogP(mol) nHBA = Descriptors.NumHAcceptors(mol) nHBD = Descriptors.NumHDonors(mol) nRTB = Descriptors.NumRotatableBonds(mol) nRC = Descriptors.RingCount(mol) mol.SetProp("MolWt",str(mw)) mol.SetProp("TPSA",str(tpsa)) mol.SetProp("logP",str(logp)) mol.SetProp("NumHAcceptors",str(nHBA)) mol.SetProp("NumHDonors",str(nHBD)) mol.SetProp("NumRotatableBonds",str(nRTB)) mol.SetProp("RingCount",str(nRC)) title = str(id) mol.SetProp("_Name",title) T1 = 'A' if mw <= 250: T1 = 'A' elif mw > 250 and mw <=300: T1 = 'B' elif mw > 300 and mw <=350: T1 = 'C' elif mw > 350 and mw <=400: T1 = 'D' elif mw > 400 and mw <=450: T1 = 'E' elif mw > 450 and mw <=500: T1 = 'F' else: T1 = 'G' T2 = 'A' if nRTB == 0: T2= 'A' elif nRTB == 1: T2= 'B' elif nRTB > 1 and nRTB <= 3: T2 = 'C' elif nRTB > 3 and nRTB <= 5: T2 = 'D' elif nRTB > 5 and nRTB <= 7: T2 = 'E' elif nRTB > 7 and nRTB <= 9: T2 = 'F' elif nRTB == 10: T2 = 'G' else: T2 = 'H' T3 = 'A' if logp <= -1: T3 = 'A' elif logp > -1 and logp <= 0: T3 = 'B' elif logp > 0 and logp <= 1: T3 = 'C' elif logp > 1 and logp <= 2: T3 = 'D' elif logp > 2 and logp <= 2.5: T3 = 'E' elif logp > 2.5 and logp <= 3: T3 = 'F' elif logp > 3 and logp <= 3.5: T3 = 'G' elif logp > 3.5 and logp <= 4: T3 = 'H' elif logp > 4 and logp <= 4.5: T3 = 'I' elif logp > 4.5 and logp <= 5: T3 = 'J' else: T3 = 'K' T4 = 'A' if nHBA == 0: T4 = 'A' elif nHBA == 1: T4= 'B' elif nHBA > 1 and nHBA <= 3: T4 = 'C' elif nHBA > 3 and nHBA <= 5: T4 = 'D' elif nHBA > 5 and nHBA <= 7: T4 = 'E' elif nHBA > 7 and nHBA <= 9: T4 = 'F' elif nHBA == 10: T4 = 'G' else: T4 = 'H' T5 = 'A' if nHBD == 0: T5 = 'A' elif nHBD == 1: T5 = 'B' elif nHBD == 2: T5 = 'C' elif nHBD == 3: T5 = 'D' elif nHBD == 4: T5 = 'E' elif nHBD == 5: T5 = 'F' else: T4 = 'G' T6 = 'A' if tpsa == 0: T6 = 'A' elif tpsa > 0 and tpsa <= 20: T6 = 'B' elif tpsa > 20 and tpsa <= 40: T6 = 'C' elif tpsa > 40 and tpsa <= 60: T6 = 'D' elif tpsa > 60 and tpsa <= 80: T6 = 'E' elif tpsa > 80 and tpsa <= 120: T6 = 'F' elif tpsa > 120 and tpsa <= 140: T6 = 'G' else: T6 = 'H' Dir_1 = T1+T2 Dir_2 = T1+T2+T3+T4+T5+T6 #fpath = 'libs2/'+Dir_1+'/'+Dir_2+'/00000' fpath = 'libs/'+Dir_1+'/'+Dir_2+'/00000' mol.SetProp("DIR_1",Dir_1) mol.SetProp("DIR_2",Dir_2) w.write(mol) isExists = os.path.exists(fpath) fname = mol.GetProp('_Name') if not isExists: os.makedirs(fpath) fname_w = Chem.SDWriter(fpath+'/'+fname+'.sdf') fname_w.write(mol) os.system("obabel -isdf "+fpath+"/"+fname+".sdf"+" -opdbqt -O "+fpath+"/"+fname+".pdbqt") os.remove(fpath+'/'+fname+'.sdf') |
16. 机器学习:重现Roy2019的BBB透过性模型
Flare V5整合了更多来源的描述符,博文《在Flare里创建血脑屏障透过性模型——自定义描述符扩展Forge QSAR建模功能》演示了如何用即将发布的Flare V5增强的QSAR建模功能,使用Flare Python拓展包RDKit计算分子描述符,建立血脑屏障(BBB)透过性分类预测模型。
17. RDKit的应用:BBB Score计算器
血脑屏障(BBB)保护大脑免受药物和外源分子的毒副作用。然而,至关重要的是,针对神经系统疾病开发的药物需以治疗浓度进入大脑。基于理化性质空间了解BBB与药物分子的相互作用可以高效地指导药物设计。Gupta等人提出一种由逐步和多项式分段函数组成的“BBB score”算法,基于五种理化描述符来预测BBB透过性:芳环的数量,重原子数,MWHBN(包括分子量的描述符,氢键供体,和氢键受体),拓扑极性表面积以及pKa。对结果进行的统计分析表明,BBB score(AUC = 0.86)优于当前采用的MPO方法(MPO,AUC = 0.61; MPO_V2,AUC = 0.67)。使用BBB score对理化性质空间的初步评估是对当前可用药物设计算法的重要补充。
我们用Juyter notebook重现了Gupta等人的BBB score计算器,使用更加方便,仅需输入SMILES代码与pKa。下载链接:https://github.com/gkxiao/BBB-score
18. 在Flare里将与某个残基相互作用的配体过滤出来
如何从1万多个Docking之后的化合物里将与某个残基相互作用的配体过滤出来? 在Flare里,可以对满足某个条件的化合物打标签(tag),然后对标签过滤。比如,我们可以将配体与残基接触做为相互作用的前提,与残基ASN253相互作用,必须先满足与残基ASN253接触。
假设配体原子与ANS253原子的距离小于3.5Å就认为有接触,那么我们可以先计算化合物与残基ASN253之间的最短距离d,如果d小于3.5 Å,则对该配体添加标签ANS253。之后,我们就可以对标签253的化合物进一步分析相互作用类型、过滤等操作。一个化合物可以有多个标签,这些标签可以组合过滤。
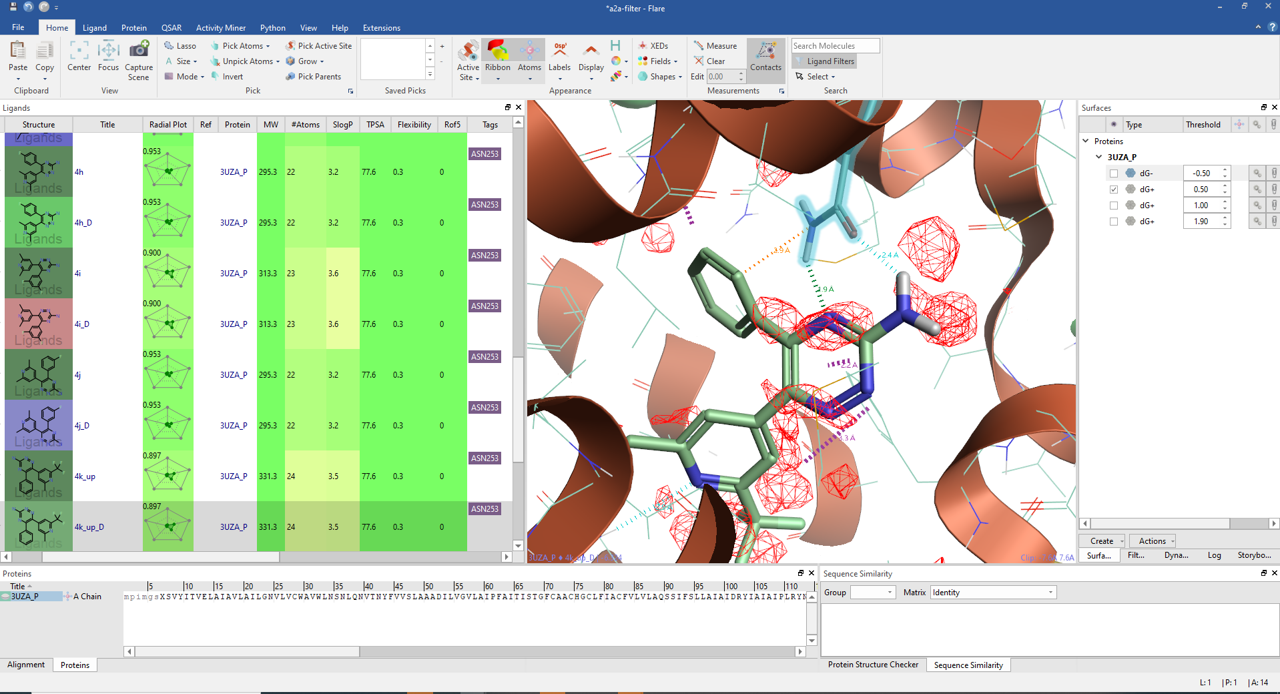
如果一个配体与残基ASN253(高亮显示 )的最短距离3.5Å,则在Tags列添加“ASN253”
如前所述Flare内置了RDKit,这里,我通过RDKit强大的Geometry来实现两个原子间距离的计算,具体方法见如下代码:
在Flare Python里执行上述代码,可以发现Ligand表单的Tag列发生变化,有的化合物多了ANS253标签。同时在Filter对这个Tag过滤,仅展示具有ANS253标签的配体。
这个方法之前用于共价抑制剂的分子对接虚拟筛选,计算Michael受体与残基Cys-SH的距离,以挑出分子对接之后满足共价反应条件的化合物,具体见原文:用常规分子对接进行共价抑制剂的虚拟筛选。
19. 从docking结果里将与某个残基发生特定相互作用的配体过滤出来
参见:分子对接后处理——与指定残基发生特定相互作用配体的过滤。以LigGrep的数据集为例,演示了如何用Flare的Python API来对与残基GLY863发生氢键相互作用、与残基TYR896发生pi-pi堆积相互作用的配体打标签,最后通过对标签的过滤,实现从虚拟筛选的结果里过滤出与特定残基发生特定类型相互作用的配体。
20. 用RDKIT解决PDBQT转SDF等其它格式文件的键级问题
以PUBCHEM的CID127化合物为例,说明如何用RDKIT的模板来归属化合物的键级。
21. 时间的加减:时长的计算
问:如何根据起始与结束时间来计算程序运行的时长?
答:用python的datetime
1 2 3 4 5 | import datetime starttime = datetime.datetime.now() # do something other endtime = datetime.datetime.now() print((endtime - starttime).seconds) |
计算两个时间点间的时长
1 2 3 4 5 6 7 8 | import datetime from datetime import timedelta, datetime, tzinfo, timezone #2022年12月8日18点06分 dt1 = datetime(2022, 12, 8, 18, 6) #2022年12月9日5点25分 dt2 = datetime(2022, 12, 9, 5, 25) #计算时长(秒) print((dt2-dt1).seconds) |
22. IC50、Ki、Kd与结合自由能ΔG的转换计算
结合自由能(Binding Free Energy, ΔG)与结合常数(Ka)解离常数(Kd)的关系如下:
$$
ΔG = -RTlnK_a = RTlnK_d
$$
其中:
- R为气体常数,值为8.314J/(mol*K)
- T为绝对温度(单位K),与摄氏温度t(°C)的关系为:T=273.15+t
- 此外,ΔG通常采用kcal/mol为单位,1 kcal = 4185.8518 J
此外有:
$$
K_d ={ 1 \over {K_a}} = {[Protein][Ligand]\over [Complex]}
$$
IC50与Ki为一半蛋白被配体占据时所需的配体浓度,此时蛋白的浓度[Protein]与复合物浓度[Complex]相等,因此有:
$$
K_d ={ 1 \over {K_a}} = IC_{50}
$$
因此有:
$$
ΔG = RTlnIC_{50}
$$
在室温25°条件下,则有:
$$
\begin{align}
ΔG & = RTlnIC_{50}\\
& = 8.314 \times (273.15+25) \times lnIC_{50} (J/mol)\\
& = 0.592 \times lnIC_{50} (kcal/mol)
\end{align}
$$
以IC50=1nM为例,在python里转换为结合自由能:
1 2 | import math 0.592*math.log(1e-9) |
得到ΔG=-12.27kcal/mol。
对结合亲和力取负对数需要用到以10为底的对数操作:
1 2 3 4 | import math # activity Ki = 5 nM, 转化为负对数pAct act=5e-9 pAct=-math.log10(act) |
23. 在Flare 配体表单插入Fsp3列
Fsp3用作描述化合物三维特征的描述符,在Flare菜单点击Python | Python Interpreter,然后复制下面的Python脚本到python解释器,点击Run按钮。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | # Welcome to Flare Python Interpreter. # Documentation can be found at Python > Documentation. # This default python script can be edited at: # 'C:\Users\Gaokeng Xiao\AppData\Local\Cresset BMD\Flare\python\default-scripts\InterpreterDefaultScript.py' from cresset import flare from rdkit import Chem p = flare.main_window().project ligs = p.ligands for lig in ligs: mol = lig.to_rdmol() fsp3 = Chem.Lipinski.FractionCSP3(mol) lig.properties['fsp3'].value = fsp3 |
计算完毕,在配体表单新增一列Fsp3。


