
摘要:用经典的分子对接方法对每个新片段组合的新产物分子进行对接打分将是非常慢而不能用于分子生长。本文详细解释了SPARK是如何解决这个问题的,并使得SPARK的Docking打分在笔记本电脑上也能够在一个晚上的时间完成对10万个分子的对接打分。
原文:Mark Mackey. 2021-09-27. https://www.cresset-group.com/about/news/optimizing-docking-spark
编译:肖高铿
用SPARK发现新的生物等排体
Spark是一种生物电子等排体替换解决方案,使药物发现研究化学家能够产生高度创新的想法来探索化学空间并摆脱IP和毒性陷阱。 它解决的问题很容易解释:“这个分子很棒! 我喜欢它! 不过,我不喜欢那一点。” 您需要对分子的一部分进行替换的原因可能有很多:物理化学或ADMET性质差、稳定性、脱靶选择性、专利问题,甚至只是您需要一个备用系列。在所有这些情况下,您都在寻找可供选择的化学结构变化,但理想情况下不会对活性产生任何显着的不利影响。Spark通过为您寻找可作为替代选择的化学物质来解决这个问题,这些化学物质可以保留您想要的性质(形状、静电和希望的亲和力),同时改变需要改变的性质(化学和可专利性)。
Spark过程的核心是寻找生物等排体。生物等排体的确切定义相当模糊,但维基百科条目解释的相当好:”生物等排体是具有相似物理或化学特性的化学取代基或基团,可产生与另一化合物大致相似的生物学特性”。 寻找生物等排体应该很容易——查看你想要替换的部分,在数据库中搜索大小和几何形状基本相同的东西,然后展示结果。 是不是很简单? 然而,如果你深入研究它,整个过程比看起来的要复杂得多。
在上下文环境下获取新的候选化合物并打分
第一个问题是如何对新的候选片段进行打分。如果我正在寻找三唑噻唑(triazolothiazole)的替代品,那么当我看到候选片段时,显而易见的事情就是问“它与三唑噻唑有多相似?”。 这种方法是有缺陷的。 原因是候选片段(和初始三唑并噻唑)的性质取决于它们的环境,即分子的其余部分。如果您对分子静电感兴趣,则尤其如此,因为分子的静电势是全局属性,通常无法分段分解。这就是Spark在产品空间而不是片段空间中进行打分的原因(图 1)。
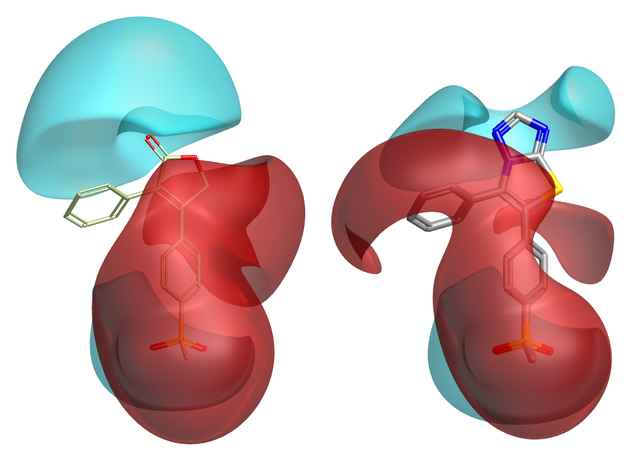
图1. 引入一个新片段后可以巧妙地改变了分子其余部分的静电:请看左边的苯基部分。
然而,即使您使用更粗略的方法(比如形状或药效团相似性)来单独处理片段也可能会产生误导,因为它可能与分子的其余部分发生空间冲突,从而彻底改变分子的构象(图 2)。

图2. 除了引起立体碰撞之外,甲基取代的苯基与苯基相似,甲基的位置还取决于R基团的性质。
为了将这些片段缝合在一起,我们还需要解决一系列其它的化学信息学与化学问题:
- 有没有原子的杂化状态发生变化?尤其对氮原子特别重要,它可以从金字塔型变为平面型。
- 是否有离子化基团的pKa发生变化?如果有,是否需要重新设置质子化状态?
- 将分子片段与其余部分缝合在一起时的静电性质是如何变化的,反过来又是什么样子?
- 如果任何新创建的键可以自由旋转,那么当新产物分子与起点分子相似性最大时的旋转异构体是什么?
- 这个构象在能量上是否合理?
解决了所有这些问题后,Spark搜索的最后阶段是对新合并的分子进行打分:它有多好? 标准Spark算法使用Cresset久经考验的静电和形状相似性算法,它可以很好地评估新片段是否确实与其替换的部分具有生物等排结构。全球数百名Spark用户都证明了这种打分算法的强大功能。
然而,还有一个应用场景还没被现有Spark工作流完全解决。有时,我们的目的不是要替换目标分子的一部分,而是希望将其往迄今为止尚未探索的结合口袋方法生长,或与蛋白发生进行新的相互作用。如果有已知的配体结合到新的口袋中,那么Spark可以使用这些配体来指导生长过程,但如果没有呢? 在这种情况下,Spark在配体引导的生物等排评估方面的出色表现是没有用的,因为没有配体可以用来指导我们。
分子对接让你可以往靶标蛋白未占据的结合口袋方向生长配体
Spark V10.6 中引入的解决方案是使用对接作为打分过程的一部分。对接打分直接由蛋白质确定,因此不再需要参考配体来告知我们活性位点特定部分的良好结合剂应该是什么样子。对我们来说幸运的是,Lead Finder对接引擎具有出色的打分算法,具有发现良好配体的能力。
优化SPARK里的对接路径
我们需要处理的主要问题是速度。Spark使用配体相似性评估片段的路径是经过高度优化的,并使用了许多经过充分测试的快捷方式,使我们能够非常快速地搜索大量片段。使用对接引擎的打分本质上要慢得多。初步测试还显示了对接方法中的一些潜在缺陷。对每个潜在的产品分子进行自由对接太慢:典型的Spark实验需要搜寻几十万个片段,而这在笔记本电脑上是不可能完成的。因此,我们将不得不依赖(就像基于配体的打分途径一样)将目标分子的母核保持在适当的位置,然后将新片段的一组预先计算的构象异构体缝合上去。显而易见的方法是使用该算法,用对接打分函数获得的分数代替配体相似性分数。然而,我们发现这并不奏效。原因是Lead Finder与大多数对接打分函数一样,对vdW相互作用的处理相当严格。仅仅将一组片段构象集合缝合到母核然后进行打分,通常会导致好的片段被丢弃,因为轻微的 vdW冲突会导致较差的对接打分。允许对接引擎在固定的结合位点里对每个pose进行优化的方法解决了这个问题,但还是太慢了——你可能有 1万个片段,但每个片段都有30个构象异构体,围绕一个新形成的单键每个可能需要用20个旋转异构体:对这些可能性中的每一种进行打分优化是不可行的。
因此,我们需要允许对接引擎对pose进行(哪怕只是轻微的)优化,但我们不可能为每个片段的所有构象异构体/旋转异构体都这样做。我们该怎么做?解决方案来自于复活了几年前的一个短期研究项目。在该项目中我们着眼于构建基于XED力场和Cresset场技术的对接打分函数。本质上,我们根据蛋白质活性位点的特性构建了一个假配体,然后使用我们现有的场/形状相似性技术将分子与该假配体叠合。当我们最初看到它时,它表现得很好,但并没有达到当时性能最好的对接引擎的标准。目前还不清楚如何弥合这一差距,因此该项目被搁置。
然而,这正是将对接引入Spark所需要的——一种非常快速地为每个候选片段选择最佳构象异构体/旋转异构体的方式,以一种容许小的立体冲突的方式,非常快速但给出了合理的结果。所以,Spark的最终算法变成了一个两阶段的事情。我们首先从活性位点构建我们的对接“伪配体”,并使用现有的非常快速的Spark算法将每个片段的一组旋转异构体和构象异构体分类为与活性位点最匹配的单个片段。然后,可以使用 Lead Finder 对最后一组结构(每个候选片段一个)进行打分,Lead Finder遗传算法允许稍微调整分子以优化其与活性位点的匹配。最终的工作流仍然比传统的基于配体的Spark实验慢,但比对所有候选产物分子进行自由对接要快得多:您绝对可以在笔记本电脑上过夜完成对数十万个片段的数据库进行 Spark对接搜索。

图3. SPARK的对接工作流(PDB: 6TCU). A) 起始分子的一部分被选中进行替换。B)删除被选中的片段。C)在截短的起始分子上以合理的取向“附”上新片段。D)用Lead Finder优化新分子的结合模式
在自己的项目中试用SPARK
对于大多数人来说,大多数时候,传统的基于配体的 Spark 实验仍然是可行的方法,它会像往常一样工作。 然而,对于那些没有指导性参比分子或不合适的时候,你真的想得到一些关于如何往你以前无法探索的结合口袋的建议,那么新的Spark对接模式正是您所需要的。
如果您想在项目中测试我们的方法,请联系我们。


