
将Flare™ Hit Expander和Flare FEP快速且准确的苗头到先导过程用于CDK9抑制剂的发现
摘要:本案例研究展示了一种联合使用取代位衍生物扫描与自由能计算的工作流,它可以围绕一个苗头化合物来快速探索周围SAR,从而加速苗头到先导(hit-to-lead)的优化过程。以CDK9抑制剂的结构优化为例,我们使用Flare(一款Cresset基于配体和基于结构的药物设计软件)中的Hit Expander模块,围绕一个起始的苗头化合物生成一系列建议,然后用Flare FEP(自由能微扰)对这些建议进行分级排序,既能从结果中检索到已知的高活性分子,又能为以提高活性为目的的进一步修饰提供明智的建议。
——联合使用取代位衍生物扫描与自由能计算来加速苗头到先导的优化过程
作者:Jenny Brookes, Tim Cheeseright, Mark Mackey
单位:Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK
编译:肖高铿/2023-04-24
前言
由于非常高的损耗率,先导化合物的发现和优化是一个漫长而昂贵的过程。经常需要合成、测试数以百计的化合物,在基于实验室的活动中花费了巨大的金钱、材料和时间。借助智能的计算方法,通过高效地优先考虑最佳分子,可以显著地帮助简化并加速这一过程。
在本案例研究中,我们展示了如何将诸如Hit Expander之类简单易用、快速的方法与准确的活性预测Flare FEP方法联合起来,然后用来可靠、快速地发现有前途的先导化合物,从而有信心地进行实验测试,节省时间和金钱。我们的研究表明,即使在感兴趣的先导系列缺乏相关晶体学数据的情况下也能获得成功。
本研究采用了美国专利US97967081公开的数据,该专利描述了氮杂吲哚类CDK9抑制剂(图1)的发明;还采用了Tong等人2后续论文公开的数据,该文描述了图2化合物的先导化合物优化过程。CDK9抑制剂很有吸引力,因为它们具有潜在的抗癌、心脏病和病毒的治疗应用前景3-5。研究还表明,它们可用于神经性疼痛、炎性疼痛与慢行疼痛的治疗6,7。
从专利中报道的1000多个抑制剂中,我们只选择了两个已知的活性化合物:一个用作建立FEP计算的参比分子;另一个作为起始的苗头化合物,用Hit Expander创建出84种变体。

图1. 专利US9796708公开的氮杂吲哚类CDK9抑制剂的通式
模拟起始的配体-蛋白复合物
化合物1(图2-左)是专利US9796708公开的CDK9有效抑制剂,IC50 = 14 nM(ΔGbinding = -10.7 kcal/mol),并表现出比CDK1、2和7高50倍以上的选择性。然而,在撰写本文时,还没有CDK9与该化合物或类似物结合的X-衍射共晶结构。因此,化合物1(4PEP)在CDK9活性位点内的结合模式是根据类似配体与CDK2的共晶结构(PDB 7M2F)建模而来,并用作计算先导优化的起点。
图2. 在本案例研究中的化合物1(也称为4PEP)来自Tong等人1的专利US9796708,对CDK9的IC50为14 nm。化合物2是CDK2共晶结构(PDB 7M2F)中的配体,作为4PEP叠合的参比化合物。
发现一个CDK9蛋白结构(PDB 6GZH)以类似的方式与强效抑制剂结合,IC50 = 11 nM(ΔGbinding = -10.9 kcal/mol)。将该蛋白叠合到CDK2的结构上,检查叠合后的结构,确认比对良好,并且配体在活性位点有相似的结合方式。然后在Flare中使用Conf Hunt & Align的配体子结构叠合方法将4PEP叠合到结合位点里,两个叠合好的蛋白都生成了相关的结合模式。CDK9蛋白是这本研究感兴趣的蛋白之一,为了评估4PEP在活性位点中模拟的结合模式的稳定性,将4PEP在准备好的CDK9蛋白(PDB 6GZH)里进行了20ns的分子动力学模拟。
初步的分子动力学模拟研究
将Molecular Dynamics (MD)作为FEP的前奏是对蛋白-配体复合物设置的有用验证,使得用户可以检查:
- 在模拟期间,感兴趣配体的结合模式是否保守。
- 配体附近的结合位点残基在电荷、互变异构体、旋转异构体上讲是否正确设置。
- 蛋白-配体和蛋白-水的相互作用是否如预期的那样发生。在本算例中,我们预期配体与CDK9的铰链区结合。
MD研究也可以帮助你确定是否需要在模型中使用二聚体/多聚体蛋白和其他可能有助于稳定的分子。对周期蛋白激酶的研究表明,一般应将周期蛋白(cyclin)包括在内。因此,对于本案例研究,由于CDK9是异二聚体复合物,因此在MD模拟中以及在后续的Flare FEP计算中均使用二聚体(由激酶和细胞周期蛋白组成)。
用3D-RISM进行水分析
由于缺乏化合物2与CDK9复合物晶体结构数据,所以对结合位点进行了仔细地水的分析。在MD运行结束时获得一个稳定的快照,然后用这个坐标在Flare中进行3D-RISM分析。本次实验使用具有先进的分子间描述的Cresset XED力场8,在有配体存在的情况下将水放置于活性位点中(图3)。然后将具有用3D-RISM预测的水位置的蛋白快照用于随后的Flare FEP计算。
使用3D-RISM来分析水的位置可能是至关重要的,因为从“正确”位置(在包埋位置或发生配体/蛋白/其他相互作用的位置)消失的水或在“错误”位置放置不当的水可能导致大的能量差异并在FEP中引入异常。使用XED力场的3D-RISM的水分析可以精确、快速地确定可能的水合位置:鉴于其简单性和非常适中的计算成本(在1-4个本地CPU上花费几分钟),在使用诸如FEP之类更昂贵的方法时值得运行该实验。
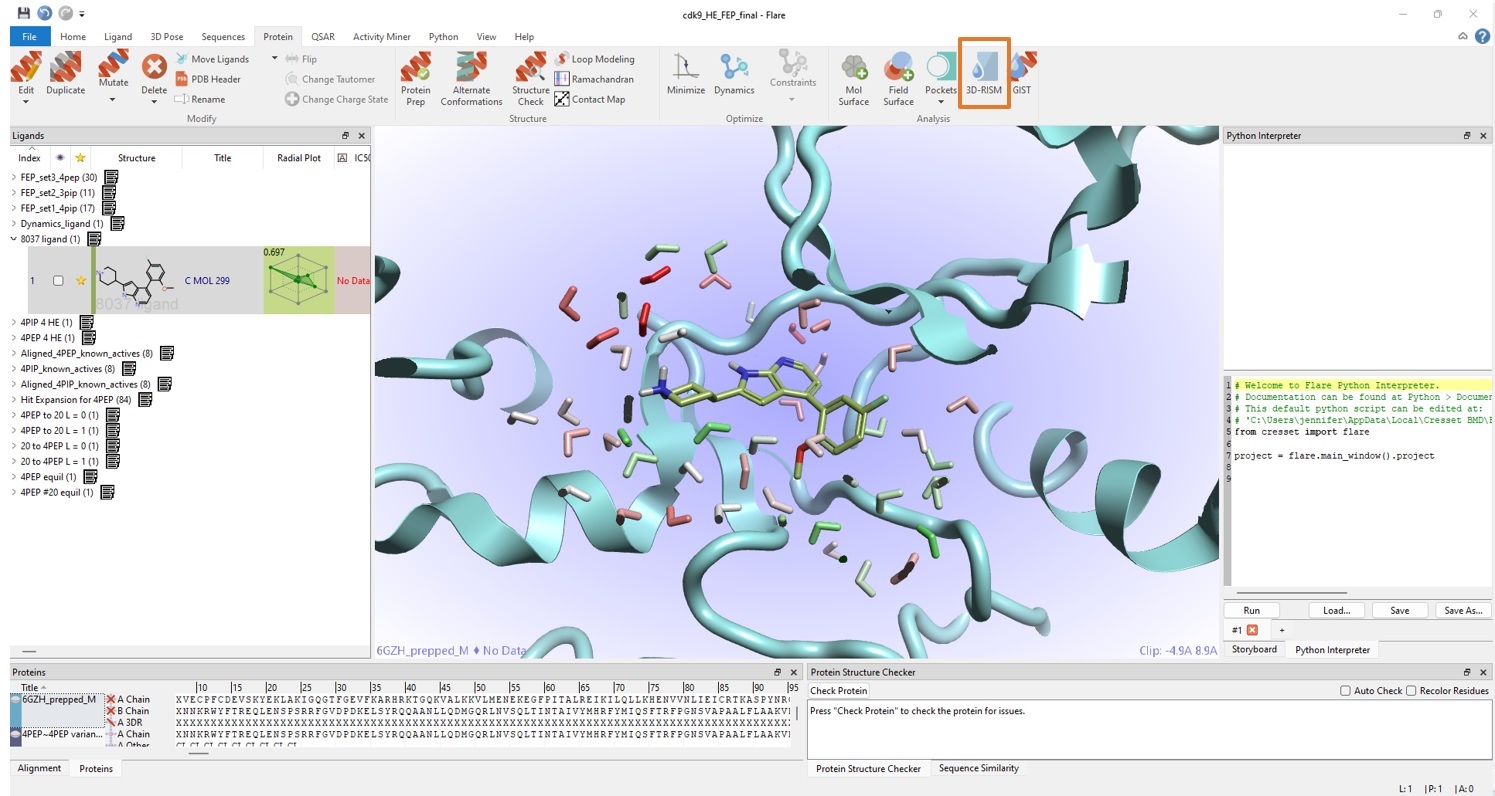
图3. Flare的3D-RISM可用于“发现”可能的水合位点,为Flare FEP模拟提供极具价值的输入。预测的水合位点的颜色从红色到绿色,分别对应于不利的ΔG和有利的ΔG。
Hit Expander
Flare的Hit Expander采用取代位衍生物扫描(positional analogue scanning ,PAS)9对苗头化合物进行细微的结构改造,从而在一个同源系列中产生了许多设计想法。在本案例研究中,我们从专利US97967081中选择了一个已知的活性化合物作为苗头化合物进行扩展。我们称之为“4PEP”(见图2的2D结构或图4-左的3D结构)的化合物显示出最小的抑制浓度和细胞抗增殖活性,对CDK9的IC50 = 24 nM(ΔGbinding = -10.4 kcal/mol),对H929细胞系的IC50 = 3000 nM(ΔG = -7.5 kcal/mol)。
在准备Hit Expander实验时,将4PEP叠合到准备好的CDK9/化合物2复合物结构上,使用了与3D-RISM实验的同一个MD快照。
在Hit Expander控制面板上还有其他选项可用,但这足以生成一大批84个苗头化合物的变体(图4-右),并迅速且充分地探索苗头化合物周围的化学景观(将在结果部分显示)。在产生了初始苗头化合物所有可能的变体后,对这些变体进行了去重复、能量最小化以产生优化的3D结构,为进一步的研究做好准备。
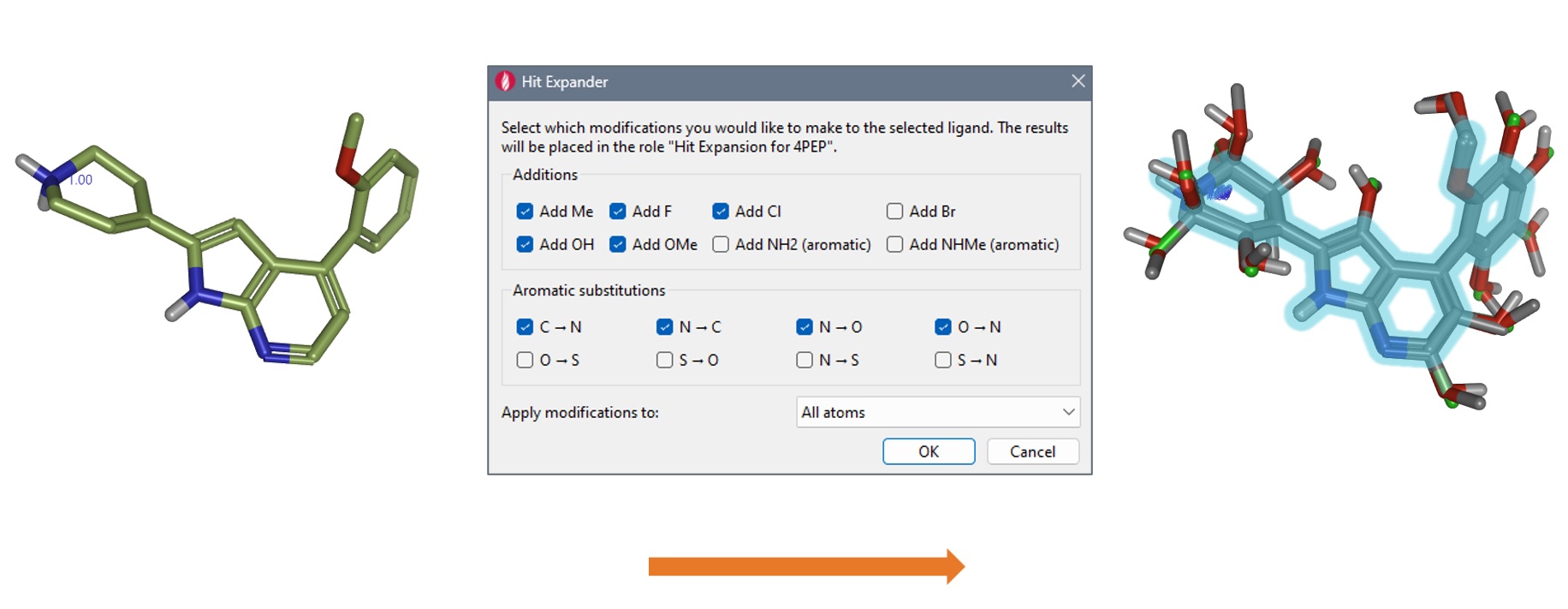
图4. 用Hit Expander(中间)对苗头化合物4PEP(左)快速地生成有前景的候选化合物。在本案例中,创建了84新分子(右)。
Flare FEP
由Hit Expander生成的苗头化合物特别适合于用相对FEP计算进行分级排序,因为所建议的分子与参比分子相比仅具有小的结构变化,并且FEP计算往往对于小的扰动更可靠。本文报道的所有Flare FEP计算采用最新的(截至撰写时)OpenFF 2.0小分子力场10,11,结合使用接近QM的半经验紧密结合方法GFN2-xTB12,13产生的定制扭转角参数,以及蛋白力场AMBER ff14SB14。
第一轮Flare FEP实验:最小跨度树,生产模式
因为只对4PEP进行了小小的改动,由Hit Expanders产生的84个变体的化合物库已经非常紧密地叠合在一起了。因此,基于已知的化合物4PEP和84个变体而建议的最小跨度树(minimum spanning tree)微扰图(图5)特别适用于第一轮的分级评估。
最小跨度树是无环、高效的拓扑结构图,其在可能的情况下最大限度地增加到中心的链接,在没有办法从中心链接的情况下则从一级链接开始分支。特别是,使用Flare生产模式(production mode)建立网络会使拓扑结构图偏好于使到达已知活性化合物的连接距离最小化(图5)。所有这些微扰都具有良好的“link score”15(大于0.6),因此可能提供可靠的预测,从而减少计算多个(冗余)连接的需求。出于这个原因,可以在Flare FEP中使用 “very Quick”模式进行FEP计算,该模式使用单向、“single mode” (从中心向外) 的微扰拓扑结构以及1ns采样时间。
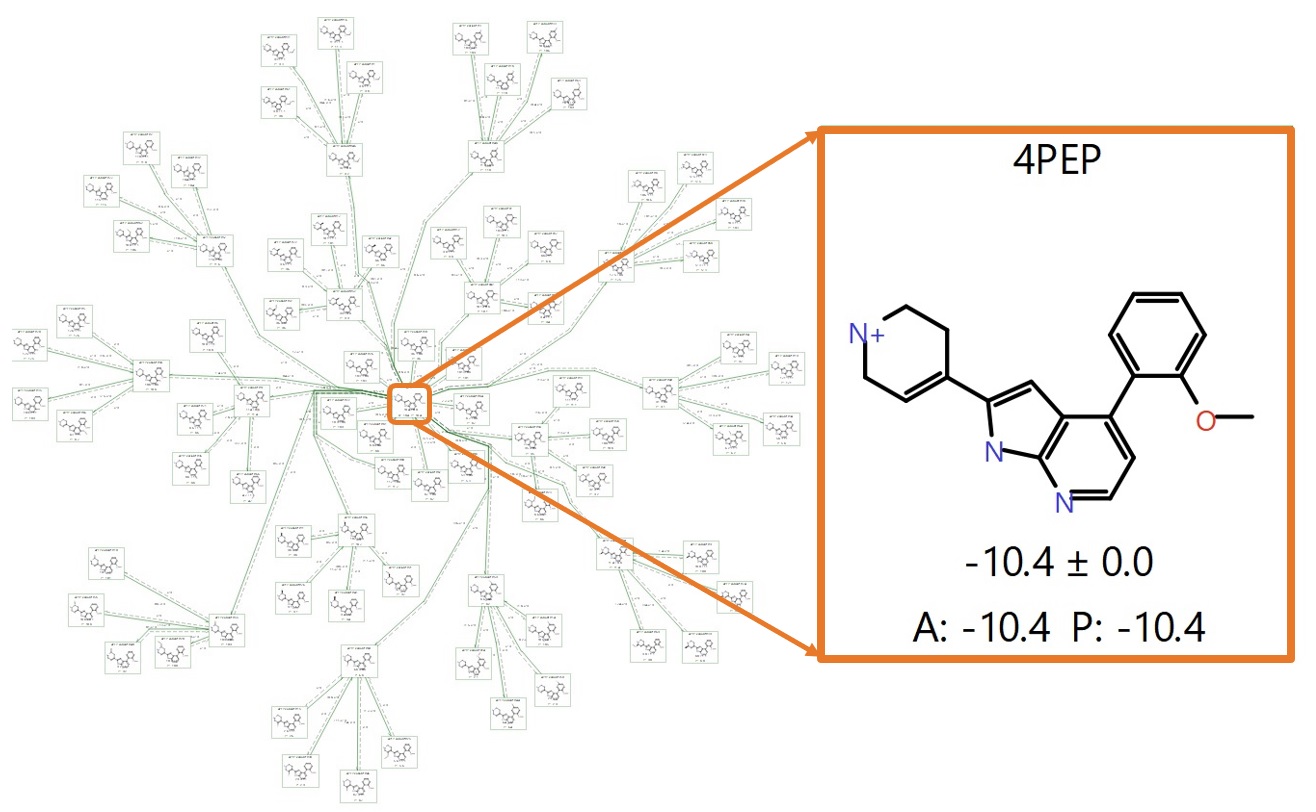
图5.以已知活性化合物4PEP(黄色方框高亮的分子)为中心,用minimum spanning tree微扰图完成的产品模式计算。A表示实际的ΔG,P表示预测的ΔG,单位为kcal/mol。
第一轮Flare FEP计算总计有85个配体,包括中心的已知活性化合物4PEP,以及84个待预测的配体,因此该图由84个链接与84个微扰组成,假设每个微扰需要使用默认9个Lambda窗口,总共有756个Lambda窗口。在实践中,Flare FEP实现了一个自适应的Lambda采样,所以收敛所需的Lambda窗口是以完全自动化的方式确定的。
第二轮Flare FEP实验:标准拓扑结构(多个连接),生产模式
第二轮FEP实验是为了完善第一轮实验的结果,对打分最高的17个结合剂(占总数的20%)进行计算。已知活性的化合物4PEP再次用作“标准”图的中心(多连接/有闭合环,图6),创建了一个有25个链接、50个微扰以及450个Lambda窗口的图。第二轮计算也是在生产模式下进行的,但是计算了两个方向的微扰,因为这提供了更大程度的误差分析并提高了总体的准确性。
在10个左右GPU的小型集群上,这两个FEP计算可以轻松地在一夜之间同时运行。在配体数据集变化不大的情况下,每个Lambda窗口仅使用1ns的采样时间就足够了。此外,在需要时,采样时间可以很容易地延长(Flare FEP默认为4ns),这可以在计算后逐个链接按需进行设定。
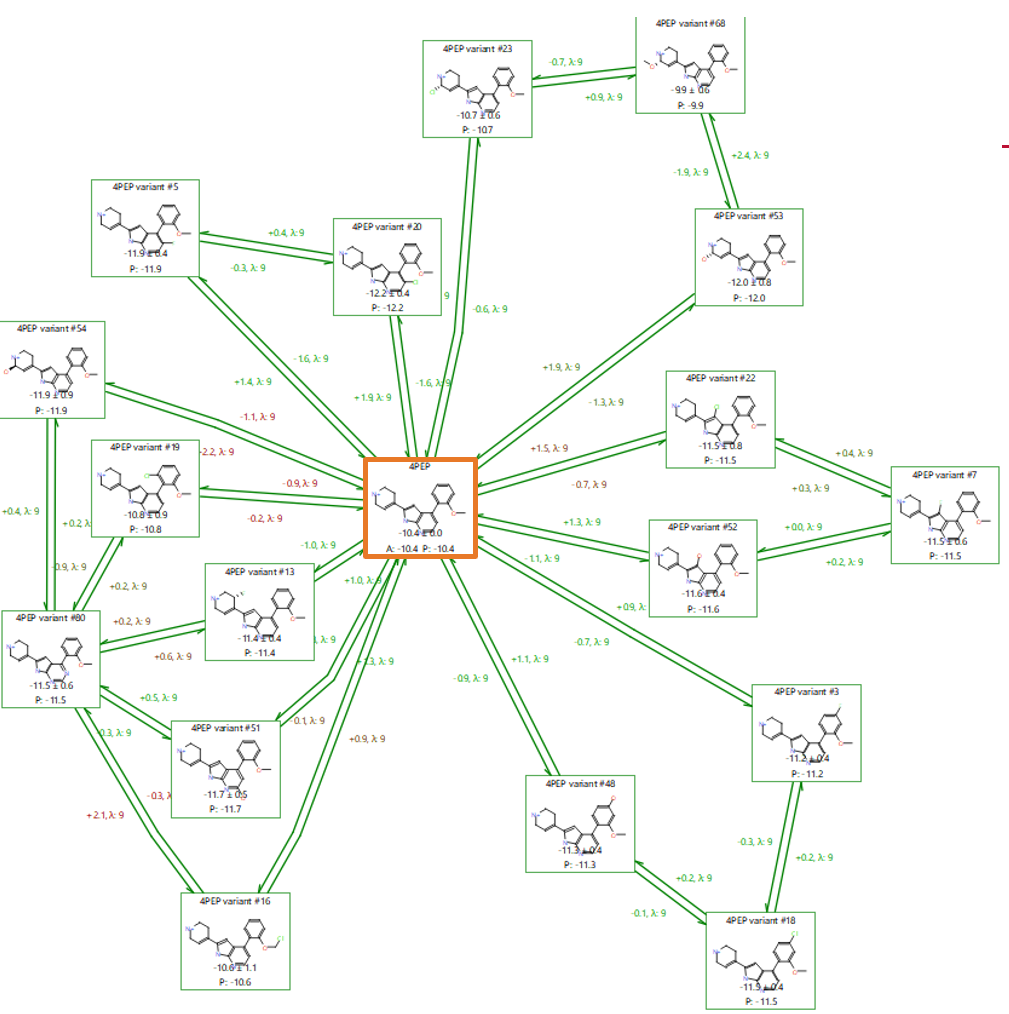
图6. 以已知活性化合物4PEP(橘黄色方框高亮显示)为中心,用标准连接图完成的Flare FEP生产模式计算
结果
标准连接图(图7)允许用户通过闭环误差分析对结果进行快速分级筛选。检查图7中的环表明,某些化合物的闭环误差较大(用橙色圈出),因此,对这些化合物的预测应被视为不太可靠。
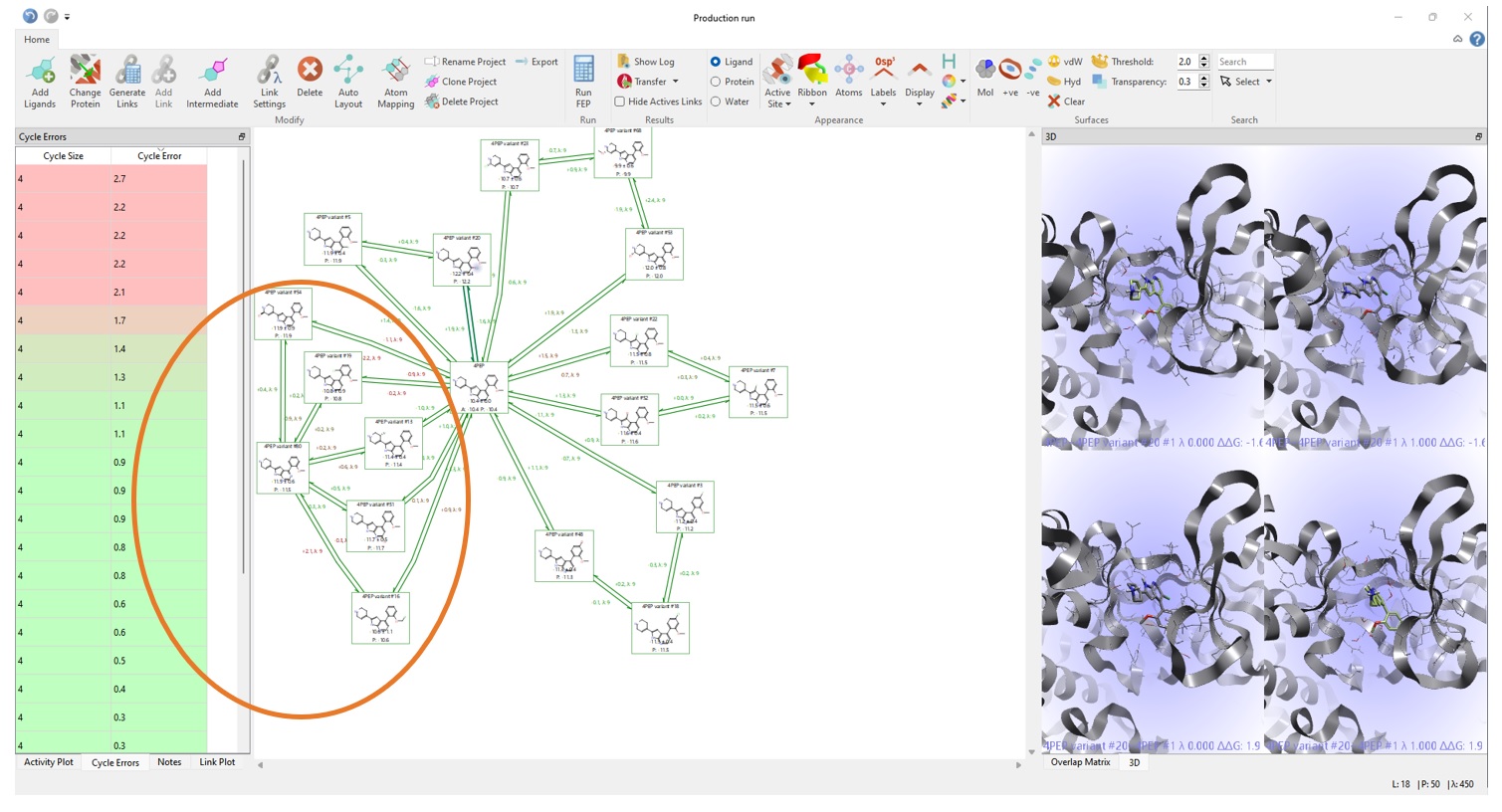
图7. 在Flare FEP项目管理中检查最终FEP结果
在图7中的两个配体(紫色方框)被Hit Expander和Flare FEP预测为良好结合剂,确实也是已知的良好结合剂。预测4PEP variant #3的结合自由能为-11.2 kcal/mol,在专利US9796708中公开的实验结合常数为16nM (ΔGbinding = -10.6 kcal/mol)。预测4PEP variant #18的结合自由能为-11.5 kcal/mol,这与实验结合常数2nM(ΔGbinding = -10.3 kcal/mol)相当。这使人们相信对其他化合物的预测也可能是准确的。
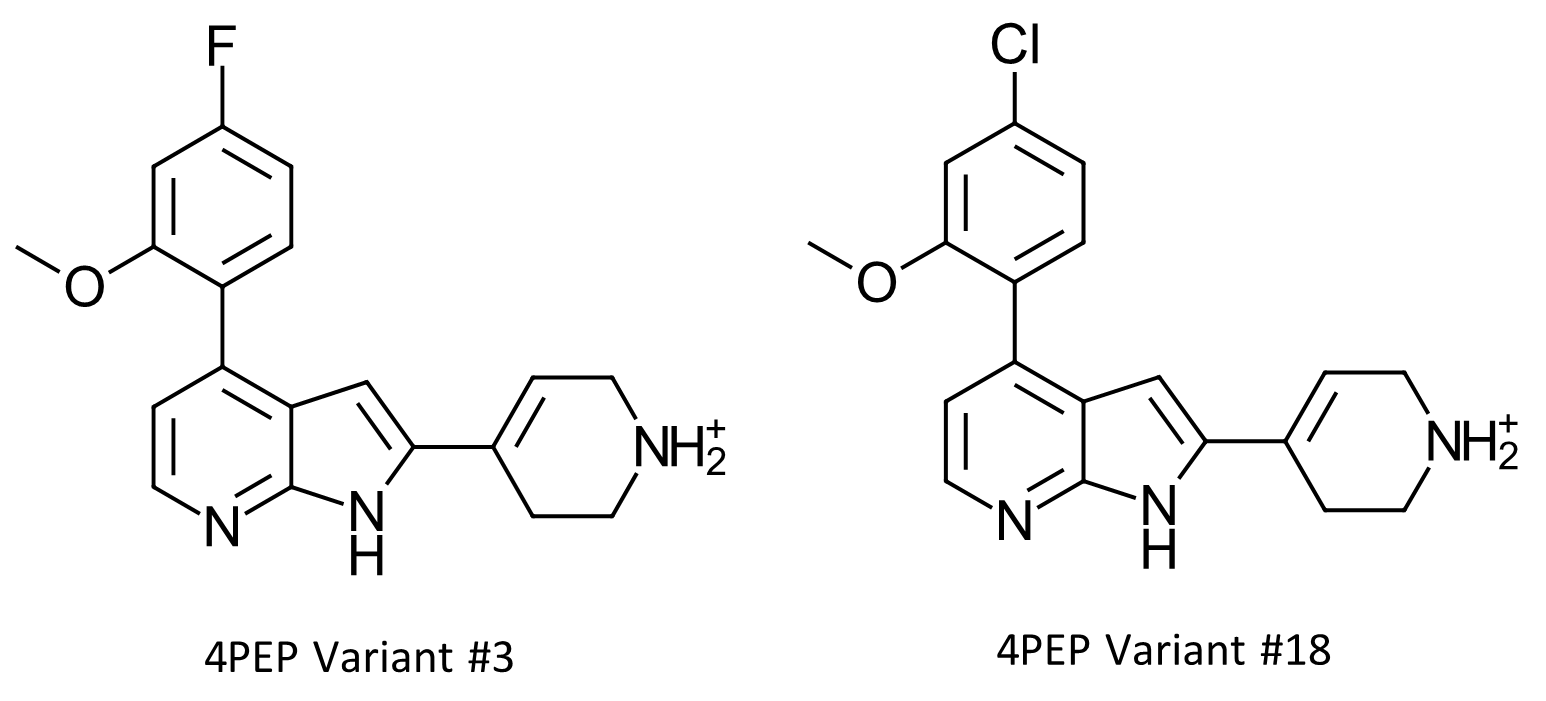
图8. Hit Expander和Flare FEP的两个设计是专利US9796708公开的化合物,并有实验的活性数据报道。Variant #3的预测ΔG值为-11.2kcal/mol,实测IC50为16 nM(ΔGbinding = -10.6 kcal/mol)。Variant #18的预测ΔG值为-11.5kcal/mol,实验的IC50值为28nM(ΔGbinding = -10.3 kcal/mol)。
除了上述几点之外,图7中的其余三个循环显示出良好的重叠、小的滞变和良好的循环误差,因此我们可以从这8种可能性中的任何一个中提出新颖的建议。然而,我们只关注图9所示的这一组循环,其包括具有最佳(更负)预测的ΔGbinding与最低预测误差的配体,在17个预测的结合剂中,4PEP variant #20排序最高(蓝色方框)。
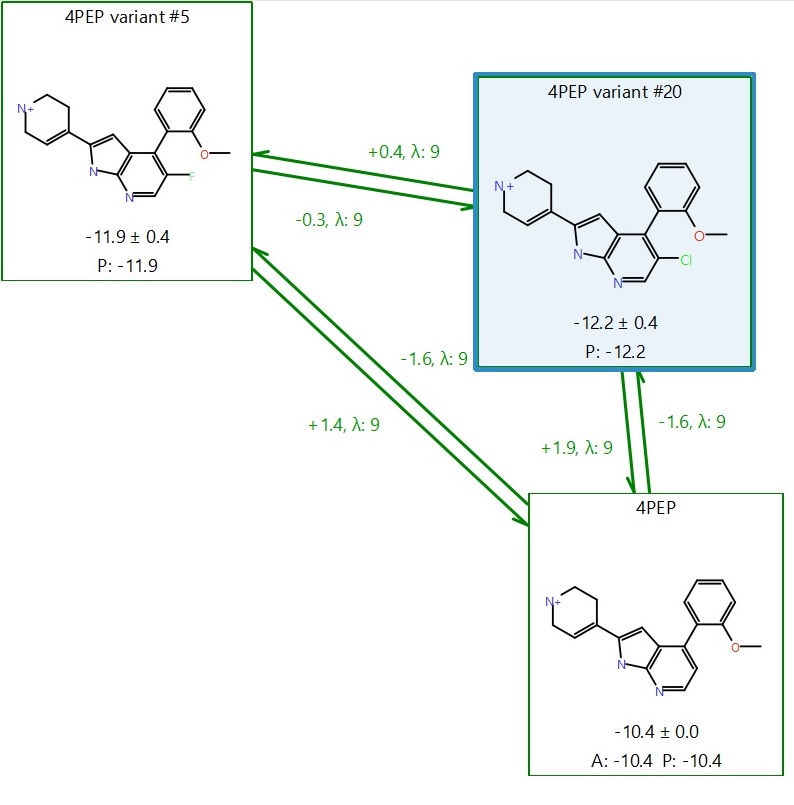
图9. 有待进一步研究的结果:先导化合物4PEP的变体variant #20(蓝色方框高亮)
图9显示了一个具有高活性预测值、低误差、前向和后向微扰之间无滞变、顺时针和逆时针方向的整体循环闭合误差ΔΔG ~ 0以及相空间矩阵中良好重叠的循环。我们可以从这里可靠地挑选出结果,并将4PEP variant #20作为潜在的先导化合物。
Variant #20预测的ΔGbinding = -12.2 +/- 0.4 kcal/mol,#20在结合自由能上有望比4PEP(ΔGbinding = -10.4 kcal/mol)好1.8 kcal/mol,这满足了这样的假设:如果FEP计算的ΔG被预测为至少比参比化合物低1.5-2 kcal/mol,那么预测的配体应该至少与参比化合物有一样的活性,或者活性更强。
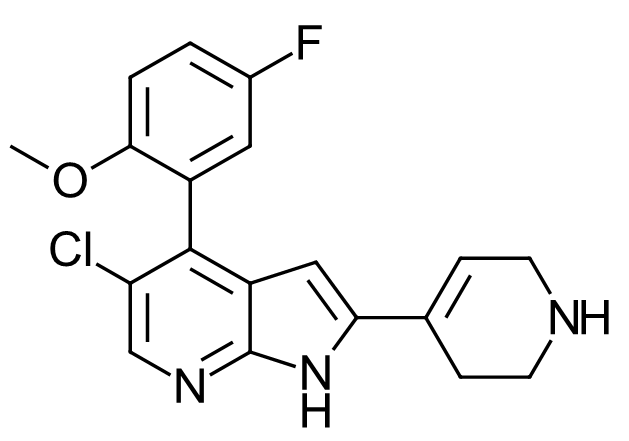
图10. US9796708专利化合物662,测得的CDK9 IC50 = 28 nM,该化合物是4PEP变体variant #20的5-氟衍生物。
虽然#20未出现在专利数据中,但一个相似的化合物(图10,US9796708实施例662)出现在专利数据中,与#20的不同之处仅在于甲氧基对位有一个氟取代。虽然该化合物的CDK9活性(CDK9 IC50 = 28 nM,ΔGbinding = -10.3 kcal/mol)比4PEP 的(IC50 = 24nM,ΔGbinding = -10.4 kcal/mol)略低,但它的细胞抗增殖活性(H929 IC50 = 83 nM/-9.6 kcal/mol)比4PEP(H929 IC50 = 3000 nM/-7.5kcal/mol)的更强。
在该专利中描述的5-位氯取代等其它变体(如#20)具有相似的趋势,具有改善的细胞生存力,可能是由于亲脂性的增加。因此,我们通过该工作流预测#20具有良好的CDK9结合亲和力并有改善全细胞数据的前景。
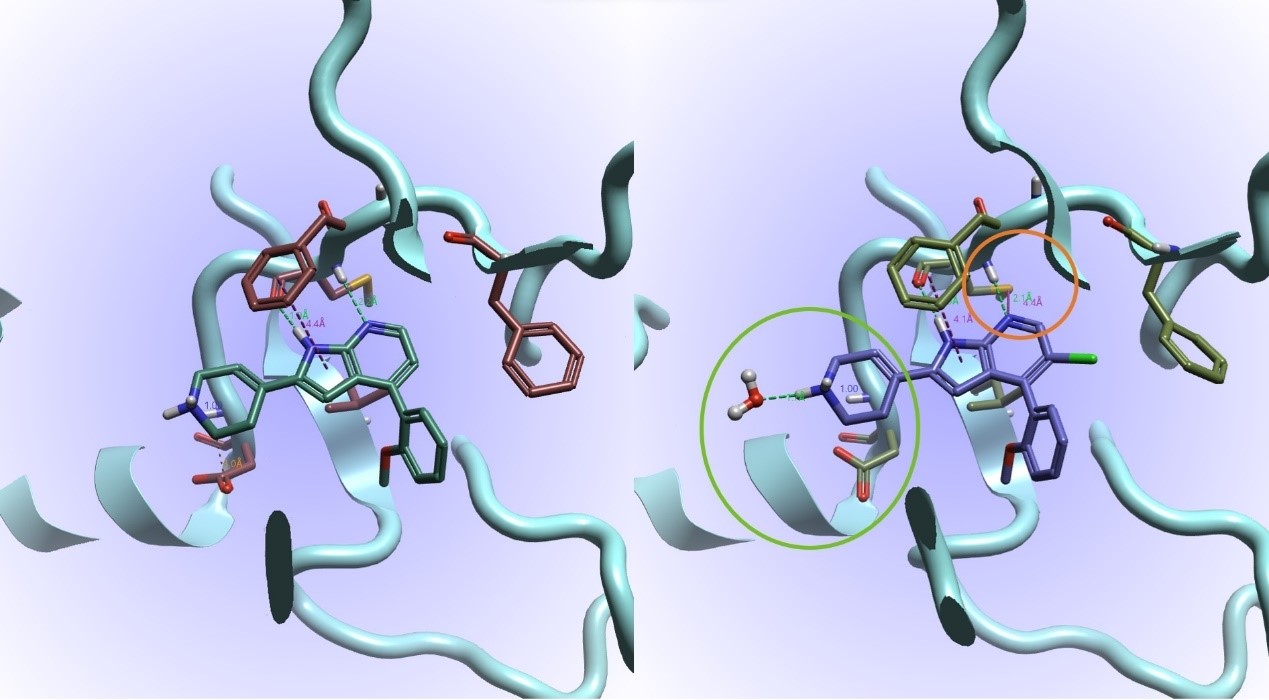
图11. 比较Flare FEP结果中平衡的4PEP(左)和variant #20(右)结构。这里显示了氢键(绿色),立体冲突(橘黄色),芳香-芳香和硫-孤对电子(均为紫色)等相互作用。
图11使我们能够在Flare FEP结果中比较平衡后的4PEP复合物(左)和先导化合物#20(右)。检查相互作用:公共的氮杂吲哚母核通过一个氢键供体与受体分别与CYS的羰基与氨基发生氢键相互作用而结合到激酶铰链上。我们可以从图9中推测,加入神奇的Cl有助于将水从疏水区挤出,并促进氮杂吲哚N上的孤对电子与CYS残基形成硫-孤对电子相互作用(橙色圈高亮),这个相互在#20中存在,但在4PEP中没有。
在#20中,氮杂吲哚相对于上述PHE有一个总体上更有利的取向,质子化的四氢吡啶与水配位,避免了与下面的ASP发生冲突(绿色圆圈高亮,水存在于#20中,而不是4PEP)。这些增强作用与#20中的邻-甲氧基保持56°扭转相结合,保持了vdW接触(已经观察到这在与CDK2的结合中很重要,PDB 7M2F2),这可能是预测为高活性的原因。
结论
在本案例研究中,我们展示了如何从一个已知结合亲和力的参比分子和一个已知活性化合物作为Hit Expander的起始分子开始,你可以迅速将一个药物发现问题从许多建议转变成几个预测,并且在使用诸如Flare FEP之类炼金术方法时可以确保高精确度。
在检查潜在的先导化合物发现时,从一个苗头化合物开发,再从84个化合物开始削减,我们重现出Tong等人报道的配体,具有已知的(良好的)活性。不仅如此,我们还发现了在CDK9结合亲和力和全细胞活性方面有可能具有更好活性的结果,并有望成为新的药物。4PEP variant #20似乎特别有希望,还有其他7种可靠的可能性值得进一步研究。
参考文献
- United States Patent Tong et al US9796708 Oct. 24, 2017 https://patents.google.com/patent/US9796708
- Tong et al. Balancing Properties with Carboxylates: A Lead Optimization Campaign for Selective and Orally Active CDK9 Inhibitors, ACS Med. Chem. Lett. 2021, 12, 1108-1115 doi: https://doi.org/10.1021/acsmedchemlett.1c00161
- Wang, S. and Fischer, PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends in Pharmacological Sciences. 2008, 29:6, 302-313. doi: https://doi.org/10.3389/fonc.2021.678559
- Alsfouk, A. Small molecule inhibitors of cyclin-dependent kinase 9 for cancer therapy, Journal of Enzyme Inhibition and Medicinal Chemistry. 2021, 36:1, 693-706 doi: 10.1080/14756366.2021.1890726
- Nomura, T., Sumi, E., Egawa, G. et al. The efficacy of a cyclin dependent kinase 9 (CDK9) inhibitor, FIT039, on verruca vulgaris: study protocol for a randomized controlled trial. Trials 20, 489 (2019). doi: https://doi.org/10.1186/s13063-019-3570-6
- Anshabo, AT. Milne, R. Wang. S. and Albrecht, H. CDK9: A Comprehensive Review of its Biology, and its Role as a Potential Target for Anti-Cancer Agents. Frontiers in Oncology. 2021, 11, 678559, 1-24. doi: https://doi.org/10.3389/fonc.2021.678559
- Hellvard, A., Zeitlmann, L., Heiser, U. et al. Inhibition of CDK9 as a therapeutic strategy for inflammatory arthritis. Sci Rep 6, 31441 (2016). https://doi.org/10.1038/srep31441
- Vinter, J. Comput.-Aided Mol. Des., 1994, 8, 653-668
- Hu, Y.; Mügge, I. In Silico Positional Analogue Scanning with Amber GPU-TI,. J. Chem. Inf. Model. 2022, 62, 18, 4448–4459 doi: https://pubs.acs.org/doi/pdf/10.1021/acs.jcim.2c00860
- Qui Y. et al. Development and Benchmarking of Open Force Field v1.0.0-the Parsley Small-Molecule Force Field. Journal of Chemical Theory and Computation 2021, 17 (10), 6262-6280. doi: https://doi.org/10.1021/acs.jctc.1c00571
- Wagner J. et al. openforcefield/openff-forcefields: Version 2.0.0 “Sage” (2.0.0). 2021. doi: https://doi.org/10.5281/zenodo.5214478
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher, S. Grimme, Extended tight‐binding quantum chemistry methods, Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11, 2, e1493.
- C. Bannwarth, S. Ehlert, S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comp. 2019, 15, 3, 1652-1671 doi: https://doi.org/10.1021/acs.jctc.8b01176
- J. A. Maier, C. Martinzes, K. Kasavajhala, L. Wickstrom, K. E. Hauser, C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 8, 3696-3713 doi: https://doi.org/10.1021/acs.jctc.5b00255
- Liu, S., Wu, Y., Lin, T. et al. Lead optimization mapper: automating free energy calculations for lead optimization. J Comput Aided Mol Des 27, 755–770 (2013). doi: https://doi.org/10.1007/s10822-013-9678-y
获取试用、商务合作,请联系我们
立即申请免费的Flare试用版,开始用Hit Expander与FEP来分析您自己的化合物系列!此外,我们还提供FTE技术服务,由我们的专家帮您完成计算任务。


