
用分子动力学模拟生成蛋白构象系综以用于生物学更加合理的分子对接实验
摘要:在本月初的网络研讨会上,我们展示了Flare中的分子动力学模拟和对接如何作为一种先进的研究技术而协同工作。在传统的对接模拟中,蛋白质固有的柔性经常被忽视,这使得它难以产生可靠的对接结合模式。在研讨会上,我们介绍了基于分子动力学模拟产生的多个蛋白质构象的系综对接工作流及其性能,本文是该研讨会的总结,希望本总结文章能帮助您探索这种功能如何支持你的研究。
作者:Matt Giblin/2023-04-03
编译:肖高铿
概述
今天早些时候,我们的应用科学家Abhijit Kayal和Ryuichiro Hara主持了一次独家在线网络研讨会,重点展示了分子动力学模拟和分子对接如何作为一种先进的研究技术协同工作。这是Flare™中的两项常用功能,Flare™是Cresset用于基于配体和基于结构药物设计的综合平台。
许多蛋白就其性质而言是柔性的,在不断变化的生理条件下会采取不同的构象。在传统的对接模拟中,这种柔性经常被忽略,因此很难为研究中的配体系列产生可靠的对接结合模式。
将一种新药推向市场可能需要十年以上的时间,耗资数十亿美元。这是科学界最复杂和最昂贵的工作之一,临床试验的失败率为90%。如图1所示,近几年来成本明显增加。然而,基于结构的药物设计技术可以使有希望的候选药物得到更有效的识别。
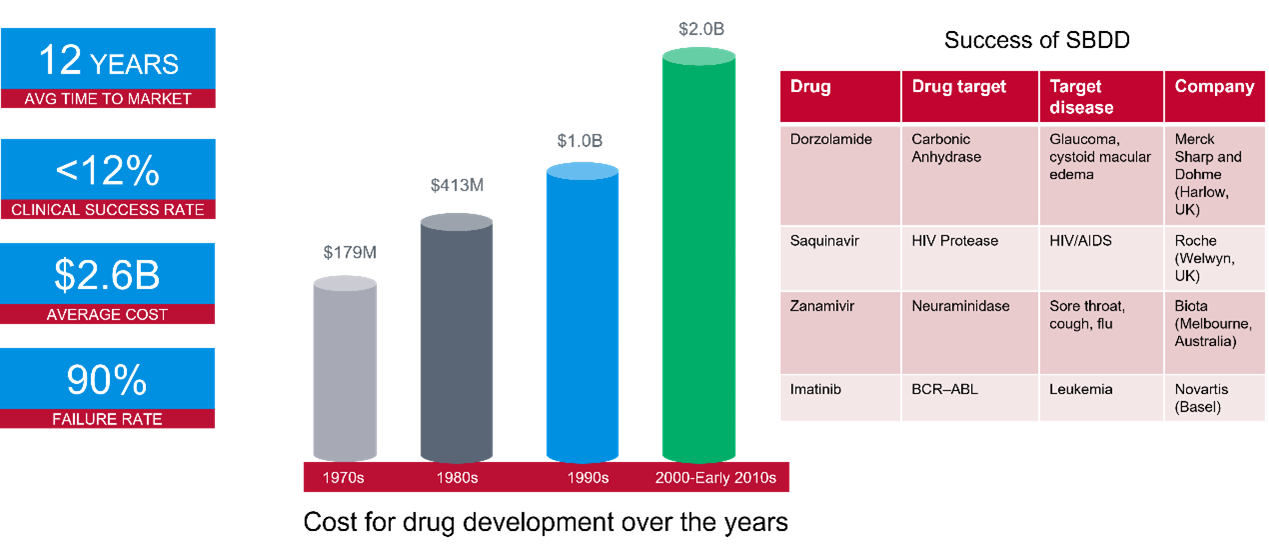
图1. 药物开发的成功率和成本,以及一些基于结构的药物设计项目实例。
对于那些不能参加网络研讨会的人,或者想了解情况的人,本文对会议进行了总结,讨论了分子动力学模拟在生成蛋白质构象系综方面所起的作用,随后的对接计算可以应用于此。继续阅读,看看这项技术如何能够显著帮助提高对接结果的准确性,并最终加速新药分子的发现。
我们在标题上标注了各个部分在网络研讨会的时间,可以通过这个链接申请会议记录。
基于结构药物设计所面临的挑战[16:40]
Abhi介绍了网络研讨会的主题,概述了蛋白-配体对接模拟所带来的一些挑战。已经有大量的打分方法可用于评估蛋白-配体复合物的结合能,每种方法有其各自独特的优点和缺点。
除此之外,基于结构的药物设计主要挑战是蛋白的柔性引起的。蛋白不是静态结构。取决于每个构象的三维结构以及活性位点的结构,不同的配体可与蛋白质的活性位点结合。例如,当活性位点处于一个扩大的位置时,较大的配体可能与蛋白的活性位点结合。不同的结合模式或结合亲和力将非常频繁地出现在不同的蛋白构象中。
用计算方法解决蛋白的柔性问题 [24:14]
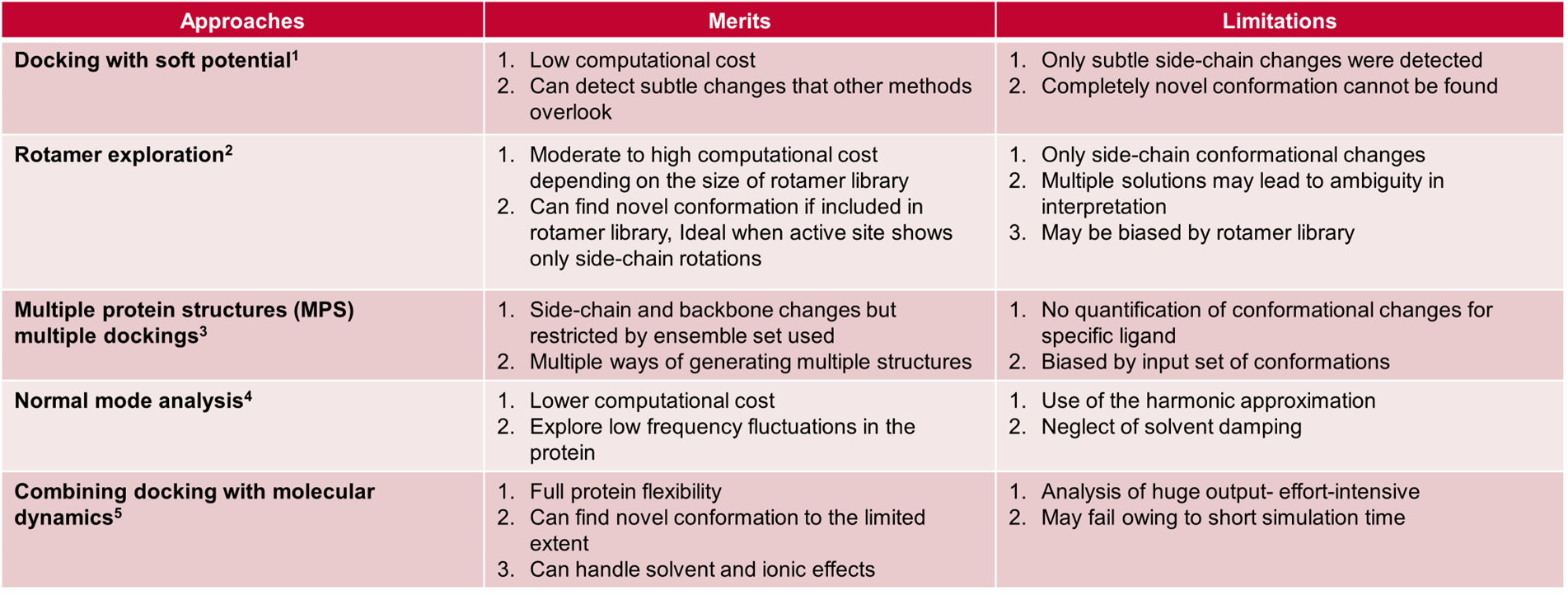
有几种不同的方法可以帮助在对接实验中将蛋白的柔性考虑在内,下面的表1对此进行了总结。
表1. 在对接实验中试用不同的方法来考虑蛋白的柔性
Cresset计算工具简介 [28:26]
Abhi接下来深入探讨了Flare的全部功能,支持用户进行优先级排序以对最佳进行合成。这套软件的流行功能包括:
- QSAR modeling
- R-group analysis
- Molecular Dynamics
- Water analysis
- Pocket analysis
- Docking and scoring
- Ligand alignment
Flare中的对接和打分使用Lead Finder™方法,它结合了遗传算法搜索和局部优化程序,高效地预测最优希望的对接模式。三种不同的打分函数可用来确定最佳结合模式的优先次序:
- Rank score: 精确的配体结合模式排序
- dG: 蛋白-配体结合能
- VS: 对虚拟筛选实验的化合物进行排序
系综对接是Flare的一项高级功能,它考虑了对接实验中蛋白活性位点的柔性。同一蛋白的多个构象可以被包括在内,并在每个构象上运行对接实验。对所有蛋白构象最佳的那个结合模式被保存为对接结果。
了解蛋白构象的动态稳定性是蛋白研究的一个基本内容,因为它在阐明蛋白与配体的相互作用和蛋白功能的机制方面起着关键作用。Flare具有创建、分析和可视化分子动力学模拟轨迹的专用界面,为研究人员提供了一个研究蛋白动态行为的强大工具。基于广泛使用的OpenMM1框架,Flare能够详细地对蛋白的构象变化及其能量进行研究,从而推进我们对蛋白的动力学和功能理解。Flare可以使用GFN2-xTB2,3扩展的半经验紧密结合或ANI-2x4,5深度学习QM近似自动生成小分子的自定义扭转参数,从而显著提高计算结果的性能。
Abhi通过以下两个案例研究展示了Flare系综对接和分子动力学模拟的协同优势。
案例 [33:55]
第一个案例研究的是CDK2(Cyclin-dependent kinase 2),在最初的自对接实验中,将两个起始配体与它们各自的共晶蛋白构象进行对接:
- 配体HMD对接到PDB 1DM2
- 配体STU对接到PDB 1AQ1
虽然这两种蛋白-配体组合都能成功自对接,但交叉对接(即试图将配体HMD对接到PDB 1AQ1)对这两个配体分子来说是失败的,表明单一的蛋白构象无法预测正确的结合模式6。
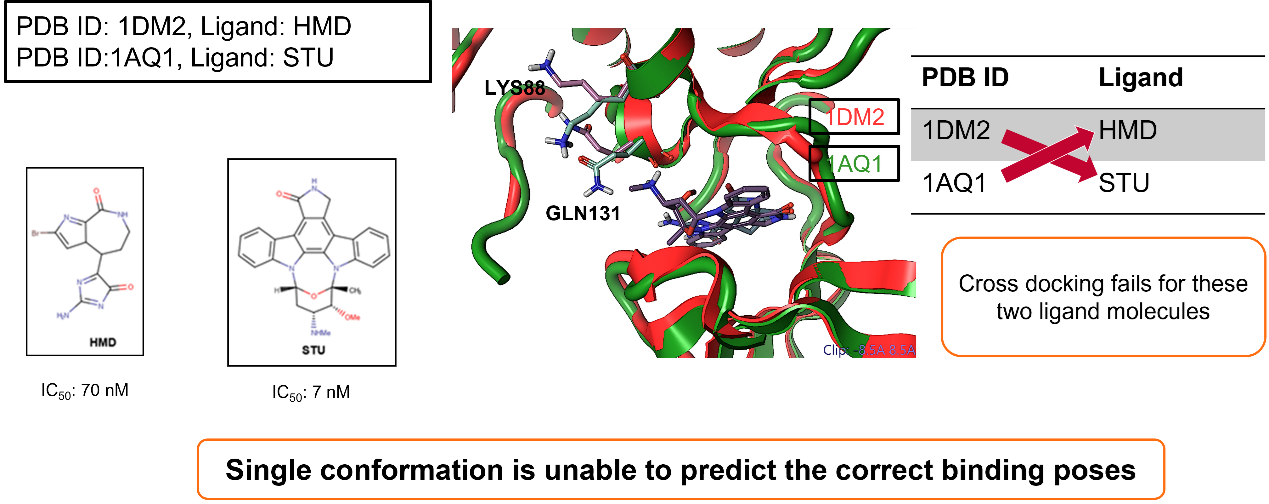
图2. CDK2对接研究案例
第二个案例研究涉及靶标Factor Xa(FXa),再次采用了两个配体,并在自对接实验中成功对接到各自的共晶蛋白构象中:
- 配体4PP对接到PDB 1XKA
- 配体FXV对接到PDB 1KSN
然而,在这个案例研究中,这两个配体分子的交叉对接也失败了,为了在对接实验中考虑多种蛋白构象需要提供了进一步的支持。
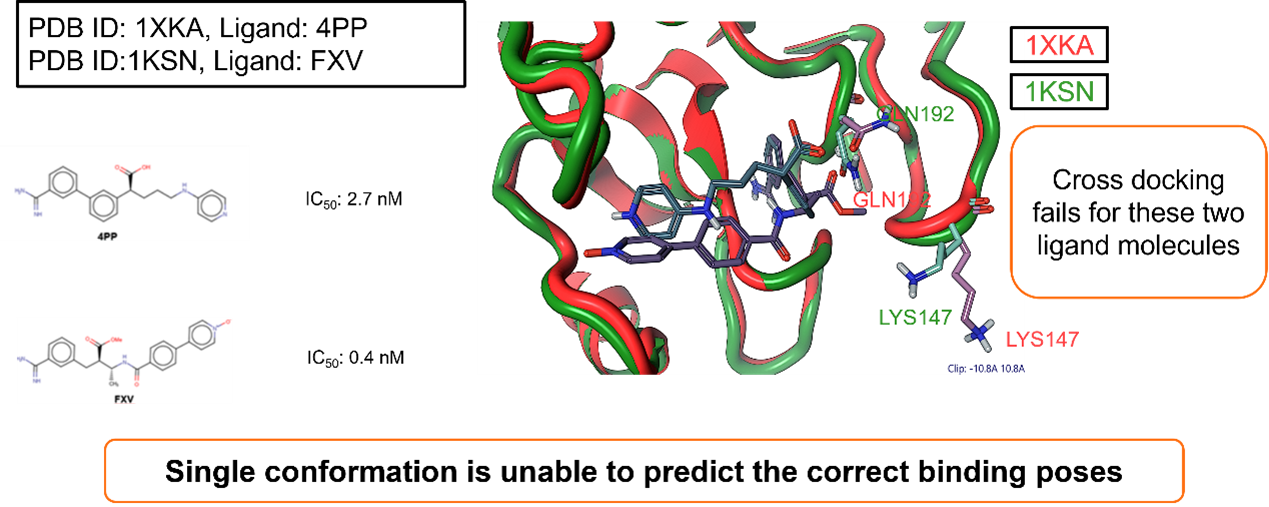
图3. Factor Xa (FXa) 对接研究案例
Cresset工作流:分子动力学模拟与系综对接[38:20]
为了在对接实验中成功地考虑多种蛋白构象,如图4所示,可以采用以下工作流:
- 从PDB获得晶体结构或用同源建模生成3D结构
- 准备蛋白结构:比如,加氢,加上缺失的loop区以获得完整的蛋白结构
- 运行分子动力学模拟:生成分子动力学模拟轨迹用于后续聚类分析,并获得多个代表性的蛋白构象
- 每个蛋白构象可后续用于系综对接实验,在对每个构象进行系综对接实验之前,需要对距离配体8Å之内的蛋白结合位点原子进行能量最小化处理。
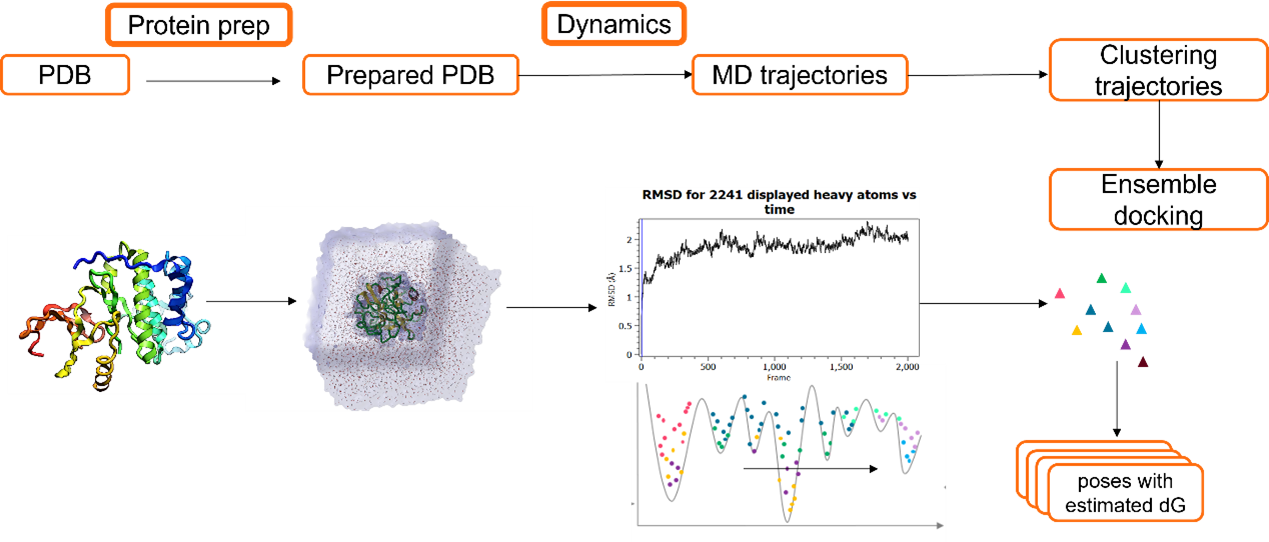
图4. 分子动力学模拟与系综对接工作流
结果与讨论 [42:35]
在图5中的两张表呈现了两个算例在原始X-ray结构上与在分子动力学模拟生成的蛋白构象系综上的自对接与交叉对接实验结果。
相比之下,采用系综对接策略的交叉对接实验产生的结合模式更接近于共晶的结合模式,预测的结合亲和力也得到显著提高。
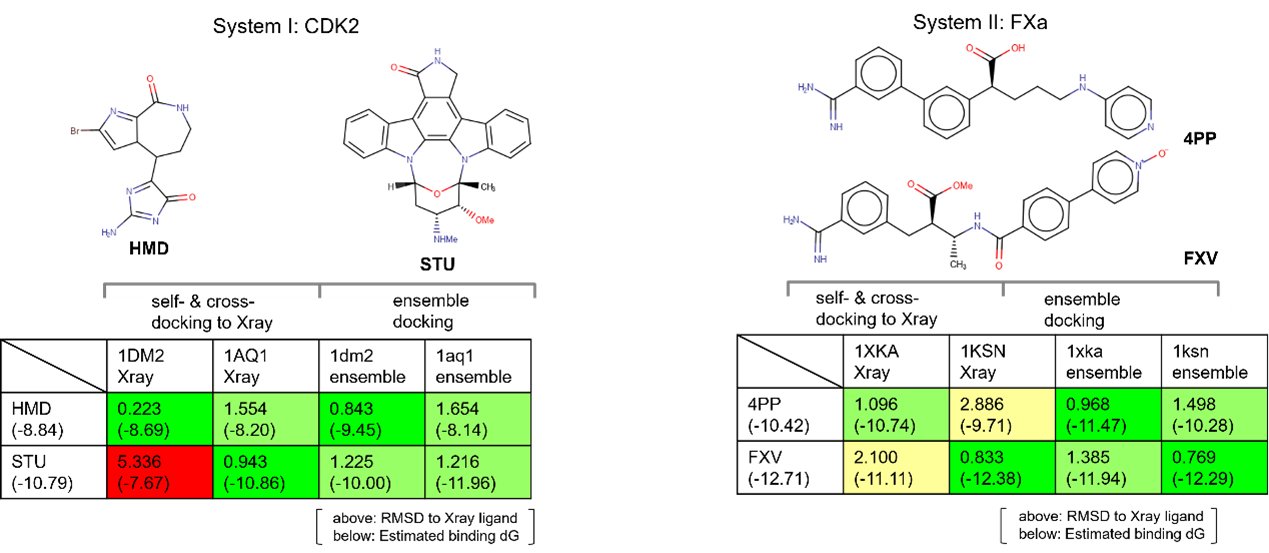
图5. 基于分子动力学模拟多构象的系综对接与基于X-Ray的自对接、交叉对接比较
结论
蛋白的柔性是自然界固有的,大多数蛋白的结合点会含有柔性的残基。因此,在用计算方法设计新配体时,考虑这种柔性是至关重要的。Flare是一个强大的工具,可以结合分子动力学模拟进行系综对接。今天就申请Flare的免费试用,以便更深入地探索这些功能以及所有其他可用的组件。
会议录像
可以通过这个页面申请获得本文讨论的网络研讨会的完整记录,以及所有其他最近的Cresset软件网络研讨会视频。
参考文献
- Eastman, P.; Swails, J.; Chodera, J. D.; McGibbon, R. T.; Zhao, Y.; Beauchamp, K. A.; Wang, L.-P.; Simmonett, A. C.; Harrigan, M. P.; Stern, C. D.; others. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS computational biology 2017, 13 (7), e1005659.
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11 (2), e1493.
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-XTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. Journal of chemical theory and computation 2019, 15 (3), 1652–1671.
- Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1: An Extensible Neural Network Potential with DFT Accuracy at Force Field Computational Cost. Chemical science 2017, 8 (4), 3192–3203.
- Devereux, C.; Smith, J. S.; Huddleston, K. K.; Barros, K.; Zubatyuk, R.; Isayev, O.; Roitberg, A. E. Extending the Applicability of the ANI Deep Learning Molecular Potential to Sulfur and Halogens. Journal of Chemical Theory and Computation 2020, 16 (7), 4192–4202.
- Campbell, A. J.; Lamb, M. L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. Journal of chemical information and modeling 2014, 54 (7), 2127–2138.
联系我们
联系我们,申请对Flare进行免费试用,以进一步探索其全部的分子模拟能力。


