
用扭转角分析解释完美的结合模式、实测活性令人失望的现象
摘要:本文用一对结合构象稳定、结合模式一致、分子对接打分区分不开活性的BAZ2A/B抑制剂5、7为例,模拟了结合模式完美、打分不比阳性参比差、但实测没有活性的场景,并用Flare QM进行扭转角分析。结果表明,不同的扭转角张力能惩罚可导致结合模式相同的分子在生物活性上的差异。在本次的计算中,用扭转角张力能校正后的结合亲和力预测值与实验值刚好一致。
前言
在虚拟筛选或结构优化时,一个比较常见的问题是:结合亲和力预测值很高、结合模式看起来完美,比之参比化合物还与靶标发生了额外的相互作用,但实测生物活性让人失望。这本质上还是探讨假阳性原因的范畴。在实践中,过高的分子内能是虚拟筛选假阳性的主要来源之一,比如在Ferreira等人1的研究中,有51%的假阳性归因于此。从理论上看,配体的构象效应不可忽视,比如早在1995年Murcko与AJay2在计算蛋白-配体结合自由能时将构象效应考虑进来。本文的主要目的是,以一对BAZ2A/B抑制剂5、7为例,演示如何通过Flare QM扭转角分析(torison profile)以发现看似完美结合模式下隐藏的构象问题,并正确理解活性差异。
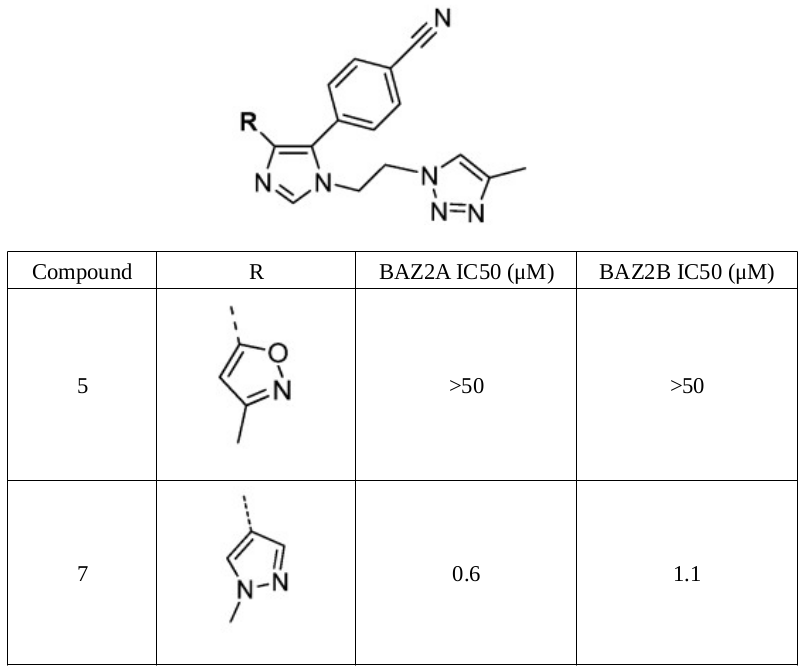
图1. BAZ2A/B抑制剂5、7的化学结构及其活性
结果与讨论
BAZ2A/B抑制剂5与7的结合模式比较
含溴结构域的蛋白BAZ2A/B在染色质重塑和非编码RNA的调节中起着至关重要的作用。Drouin等人3从弱活性起点分子出发,通过快速优化,基于结构发现了一种有效的、选择性的、细胞活性的BAZ2A/B溴结构域抑制剂BAZ2-ICR。在此过程中,合成了一对结构相似的抑制剂5、7,如图1所示,其中5没有显著的活性,但是7具有低μM水平的活性。
如图2-左的7与BAZ2B的共晶结构PDB 4XUB表明,甲基吡唑环氮原子可与ASN1994以及结合水HOH2199发生氢键相互作用。用Flare V84在结合口袋里对7进行编辑、用甲基异噁唑替换甲基吡唑之后得到化合物5的结合模型,如图2所示,可发现5与BAZ2B的相互作用与7一致;此外,5的甲基异噁唑氧原子还可以与HOH2112发生额外的氢键相互作用,这让人期待5的活性至少不低于7。
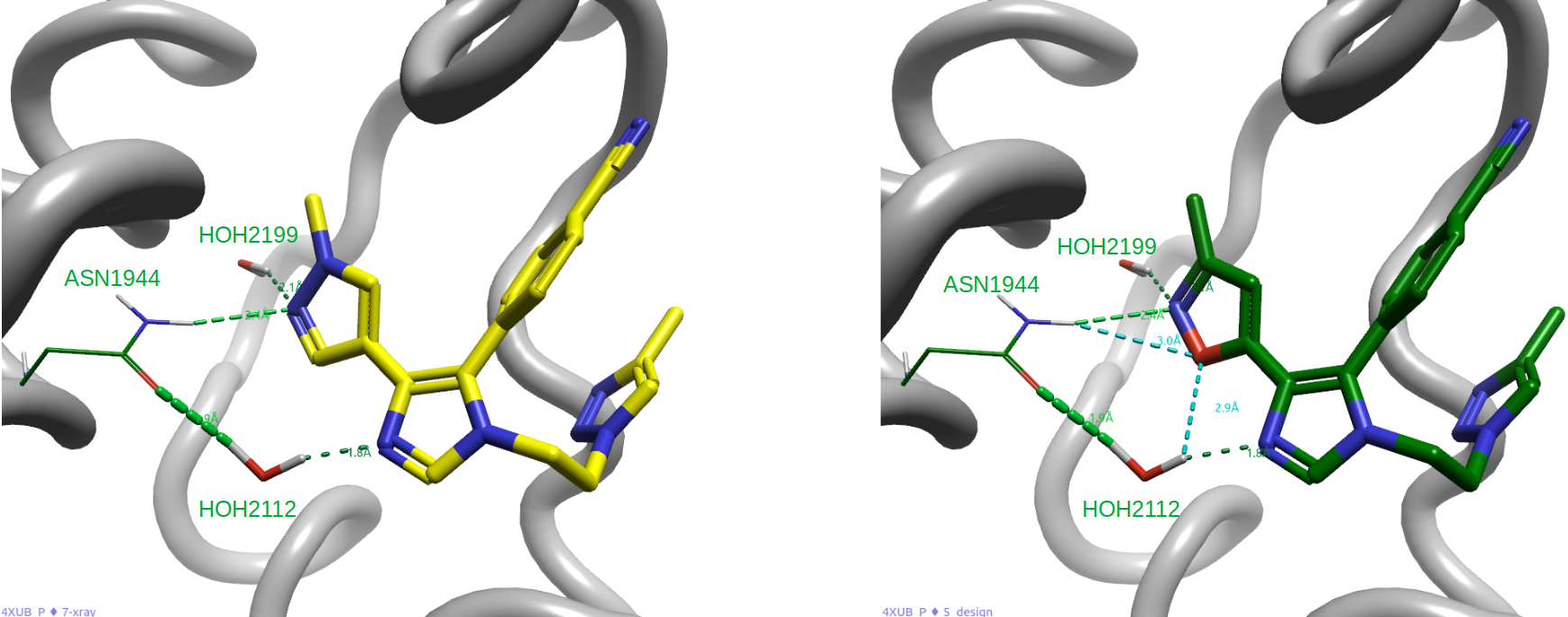
图2. 化合物5、7的结合模式。左:7的结合模式(PDB 4XUB);右:5的结合模式(根据7建模)。灰色飘带:蛋白结构。
用Flare QM4在BP86-D3BJ/DEF2-TZVP理论水平下对化合物5、7进行最小化计算,结果发现优化后的构象与X-Ray构象之间的RMSD均小于0.3Å,最大原子间距离为0.5Å,因此认为它们的生物结合构象就是能量极小点,不存在诱导契合引起的配体构象焓变,这与以往的Torsion profile算例显著不同。
用分子对接预测BAZ2A/B抑制剂5与7的结合亲和力
AutoDock Vina是一款流行的分子对接软件5,GNINA是Vina的深度学习打分版本6,7。用GNINA对化合物5、7进行score_only模式对接计算,计算时将HOH2112、2199作为受体的一部分,结果如表1所示,输入与输出文件见附件。
表1. 分子对接GNINA打分结果与实验活性值
| Items | 5 | 7 |
|---|---|---|
| gauss 1 | 79.64332 | 80.71584 |
| gauss 2 | 1187.72916 | 1187.06944 |
| repulsion | 2.74163 | 2.85858 |
| hydrophobic | 21.98071 | 15.07247 |
| H-Bond | 4.06525 | 3.79043 |
| Affinity score (kcal/mol) | -7.26 | -6.92 |
| CNNaffinity(pKd) | 6.4 | 6.6 |
| CNNaffinity(ΔG kcal/mol)a | -8.7 | -9.0 |
| CNNscore | 0.92 | 0.97 |
| BAZ2B IC50 (μM) | \( >50 \) | 1.1 |
| BAZ2B ΔG (kcal/mol)a | \( >-5.86 \) | -8.12 |
注:AutoDock Vina的打分项为加权之前的值。a:根据亲和力或IC50换算得到。
首先注意到Vina score的氢键打分项,5比7高了0.3这反映了5的甲基异噁唑环氧子与ASN2199、HOH2112之间比7额外的氢键相互作用;其次,5比7具有稍微略高的输水相互作用项。这两项导致5比7的Vina score略优,结合自由能预测值分别为-7.26与-6.92kcal/mol,没有显著的差异。相似的,GNINA的深度学习CNNaffinity对5、7结合亲和力预测值(pKd)分别为6.4与6.6,也没有显著差异。这说明,无论是经典的Vina score还是最近开发的深度学习亲和力打分都不能区分5与7在活性上的差异。
GNINA的CNNscore是评估pose为正确结合模式的概率,5与7的CNNscore均大于0.8,这说明虽然结合亲和力预测值不能对化合物排序,但是结合模式是正确的概率相当大。我们还可以发现,CNNaffinity对7的预测值为6.6(pKd),这与实验值IC50=1.1μM相当。
扭转角分析:对结合亲和力预测值施以构象效应的校正
用Flare QM在BP86-D3BJ/DEF2-TZVP理论水平下对5、7的模型分子进行扭转角分析,结果如图3所示,5、7的生物活性构象(两面角=-22°)张力能并非全局最低能构象,它们的张力能分别是3.35、0.83kcal/mol。这说明,在与BAZ2B以同一个模式结合时,5受到的惩罚比7的大了约2.5 kcal/mol,这可用来解释为什么5的体外活性比7的差。
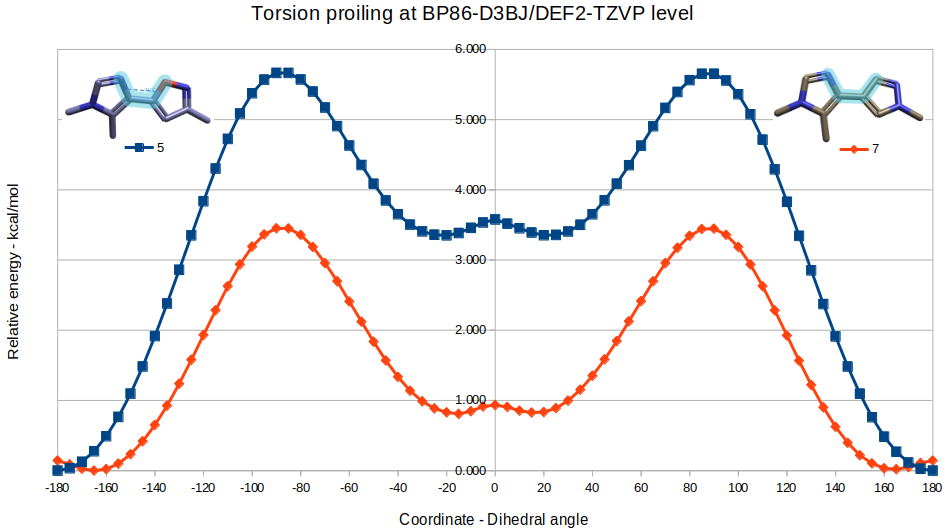
图3. 化合物5、7模型分子的扭转角分析。高亮的原子:定义扭转角分析的两面角。
同时,5、7的生物活性构象虽然不是全局极小点,但却是一个能量极小点,因此不存在因诱导契合而使得两面角偏离极小点,进而产生结合构象焓升高的情况。这与之前的QM最小化计算结果是一致的。
由于5、7之间的区别仅在于甲基异噁唑与甲基吡唑片段,因此可以假设活性差异仅由这两个片段与咪唑之间可旋转键的扭转角张力能差异引起的。鉴于具有高的CNNscore,以及CNNaffinty可以较准确地预测7的亲和力,因此值得尝试按照Murcko与AJay2的方法,用计算的5、7扭转角张力能(Strain energy,ΔE)对CNNaffinty进行校正,也就是:
$$CNNaffinity\_corrected = CNNaffinity + ΔE$$
用扭转角张力能校正后的CNNaffinity结果如表2所示,化合物5、7校正后的结合自由能预测值分别为-5.37、-8.17kcal/mol,这与它们的实验值\(>\)-5.86、-8.12kcal/mol是一致的。当然,这很可能只是一个巧合,但是可以说明在打分函数CNNaffinity里并没有考虑构象张力能,这需要我们额外加以考虑。
表2. 用扭转角分析张力能校正结合亲和力预测值
| Items | 5(kcal/mol) | 7(kcal/mol) |
|---|---|---|
| CNNaffinity(ΔG)a | -8.7 | -9.0 |
| ΔEb | 3.35 | 0.83 |
| CNNaffinity_corrected | -5.37 | -8.17 |
| BAZ2B ΔGa | \( >-5.86 \) | -8.12 |
a. 根据亲和力或IC50换算得到;b. 扭转角分析的张力能。
总的来说,假设5、7具有相同结合模式的情况下,扭转角分析表明5、7的生物活性构象虽然都为能量极小点,但不同的扭转角张力能惩罚可导致5、7的活性差异。对此可以用扭转角张力能解释活差异,并用扭转角张力能校正的CNN结合亲和力打分预测化合物的生物活性。
材料与方法
蛋白结构与配体结构的准备
将7与BAZ2B的共晶结构PDB 4XUB下载到Flare V8,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,并对截短的蛋白质链继进行封端。从准备好的复合物结构里将配体提取出来,得到7的生物活性构象与蛋白结构。在Flare里复制配体7,并将甲基吡唑用原子替换的方式继续编辑,得到化合物5,并进行能量最小化。
QM几何优化
QM几何优化使用Flare QM的Minimize在BP86-D3BJ/DEF2-TZVP理论水平下计算。
模型分子的准备
模型分子是从上一步准备好的活性构象配体基础上截取得到,截小后的模型分子保留其构象与生物活性构象一致,并作为后续Torsion Profile分析的起点。
QM扭转角分析(Torsion Profile)
用Flare QM的Torsion Profile在BP86-D3BJ/DEF2-TZVP理论水平下对模型分子的两面角进行扭转角分析,计算参数如下:
- Method: DFT
- DFT functional: BP86
- Use dispersion correction:Yes
- Basis set: def2-tzvp
- Convergence: Medium
- Max iterations: 500
- Number of rotomers: 72
- Degrees: 5.0°
- Max Threads: Auto
其中Degrees是根据Number of rotomers自动生成,无需主动设置,这里意味着以5.0°为步长进行两面角扫描。
结论
本文用一对结合构象稳定、结合模式基本一致、分子对接打分区分不开活性的BAZ2A/B抑制剂5、7为例,模拟了结合模式完美、打分不比阳性参比差、但实测没有活性的场景,并用Flare QM进行扭转分析。结果表明,不同的扭转角张力能惩罚可导致结合模式相同的分子在生物活性上的差异。在本次的计算中,用扭转角张力能校正后的结合亲和力预测值与实验值刚好一致。
附件
分子对接输入与结果文件:baz2b_docking.tar.gz
1 2 3 4 5 6 7 8 9 | baz2b_docking/ ├── 4xub_prot.pdbqt ├── 5_out.sdf ├── 5.sdf ├── 7_out.sdf ├── 7.sdf └── dock.conf 0 directories, 6 files |
参考文献
- Ferreira, R. S., A. Simeonov, et al. Complementarity between a docking and a high-throughputscreen in discovering new cruzain inhibitors. J Med Chem, 2010, 53(13):4891-4905. https://pubs.acs.org/doi/10.1021/jm100488w.
- Murcko, Mark A. et al. Computational Methods to Predict Binding Free Energy in Ligand-Receptor Complexes. Journal of Medicinal Chemistry, 1995, 38(26), 4953–4967. doi:10.1021/jm00026a001
- Drouin, L. et al. Structure enabled design of BaZ2-ICR, a chemical probe targeting the bromodomains of BaZ2a and BaZ2B. Journal of Medicinal Chemistry, 2015,58(5),2553–2559. https://doi.org/10.1021/jm501963e.
- Flare V8. https://www.cresset-group.com/software/flare
- Trott, O. and Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 2009, 31(2), 455–461. Available at: https://doi.org/10.1002/jcc.21334.
- Sunseri, J., & Koes, D. R. Virtual Screening with Gnina 1.0. Molecules, 2021, 26(23), 7369. https://doi.org/10.3390/molecules26237369
- GNINA 1.0, https://github.com/gnina


