
摘要:AMBER力场常用于分子动力学模拟与FEP计算,虽然GAFF力场对大部分体系计算精度足够,但是对于某些分子却不能良好地描述。因此有必要自定义力场参数以提高计算精度。本文以Imatinib为例,详细介绍了如何用Flare的自定义力场功能优化小分子的力场参数并用于后续的分子动力学模拟与FEP计算。
作者:肖高铿/2020-12-17
1 背景
分子力场是分子力学计算的基础,精确的构象搜索、分子动力学模拟与炼金术法FEP计算结合自由能等方法都依赖于精确的力场。AMBER力场是常用的分子力场,通常用其中的GAFF/GAFF2来描述小分子的原子间相互作用。虽然GAFF/GAFF2力场适合于大部分体系,然而对某些体系并不适合,此时就需要自定义力场参数。
自定义力场参数的第一步是选择需要改进描述的两面角;然后用QM对选定的两面角进行扫描;最后拟合新的力场参数以便用于后续的计算比如分子动力学模拟与FEP计算。两面角选择可以根据来自前期AMBER力场报告中的提示、分子动力学模拟或FEP计算结果分析发现的不合理构象相关的可旋转键。然后对感兴趣的可旋转键进行基于QM的两面角扫描得到每个两面角的相对构象能量。在Flare里,内置了PSI4程序用于QM计算。Flare除了AMBER力场之外,还有Open Force Field(OFF)力场,但是目前的自定义力场参数不支持OFF。
2 自定义力场算例
2-苯氨基嘧啶(图1高亮部分)是激酶抑制剂中常见的片段,比如上市药物Imatinib(图1,左)与Brigatinib(图1,右)即包含这个片段。本文以该片段的两面角ca-nu-ca-nb来说明如何用Flare自定义力场参数,并用于后续的分子动力学模拟或FEP计算。
|
|
|
图1. Imatinib与Brigatinib的化学结构
3 自定义力场的详细过程
3.1 打开自定义力场向导
在“菜单|Custom Force Field”下拉菜单点击Custom Force Field,如图2所示。此时弹出Custom Force Field Wizard对话框(图3)。
图2. 打开Custom Force Field向导
3.2 选择需要生成参数的力场
在Custom Force Field Wizard对话框(图3)中,点击需要生成参数的力场。在本次练习中,我们点击GAFF2。
图3. Custom Force Field向导
3.3 选择两面角
在配体3D视窗区用鼠标点击组成两面角的四个原子(图4),或者在Recommended Torsions下拉菜单(图4)中选择两面角。
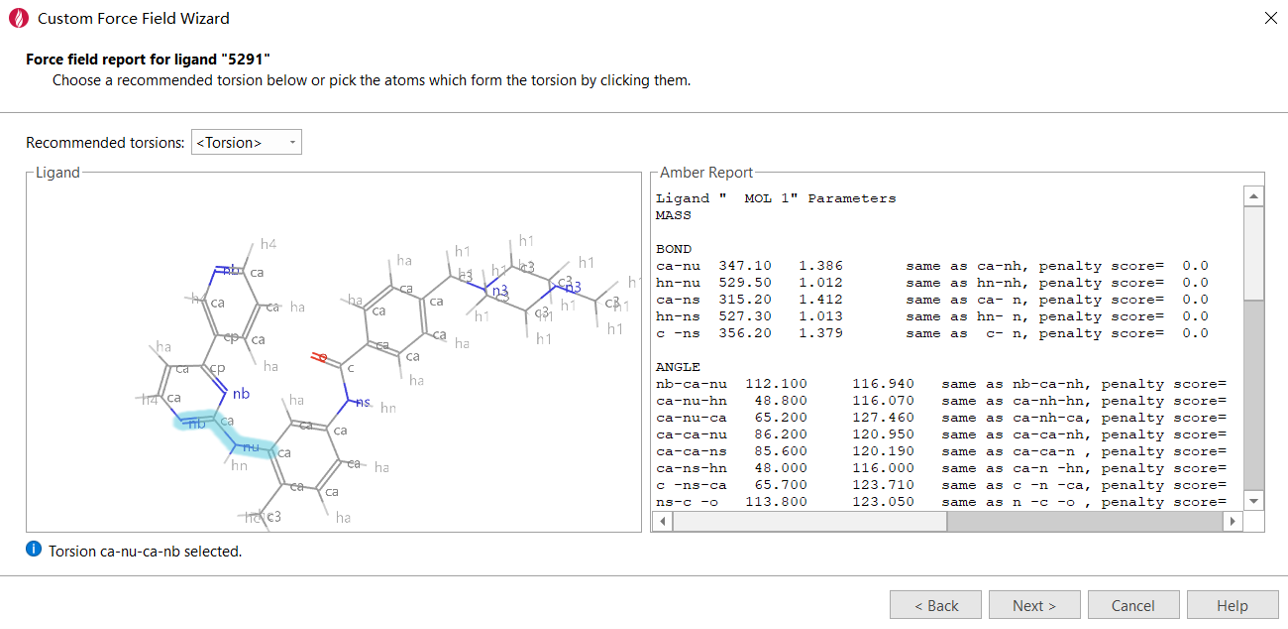
图4. 选择两面角
选好两面角后,然后点击next按钮,弹出创建代表性片段对话框(图5)。
3.4 创建代表性片段
我们会看一个黄色三角的警告提示:片段的重原子数大于20个、3个可旋转键,为了节省计算时间可以考虑将分子裁小。我们选择对分子进行裁剪,点击Trim Ligand按钮。
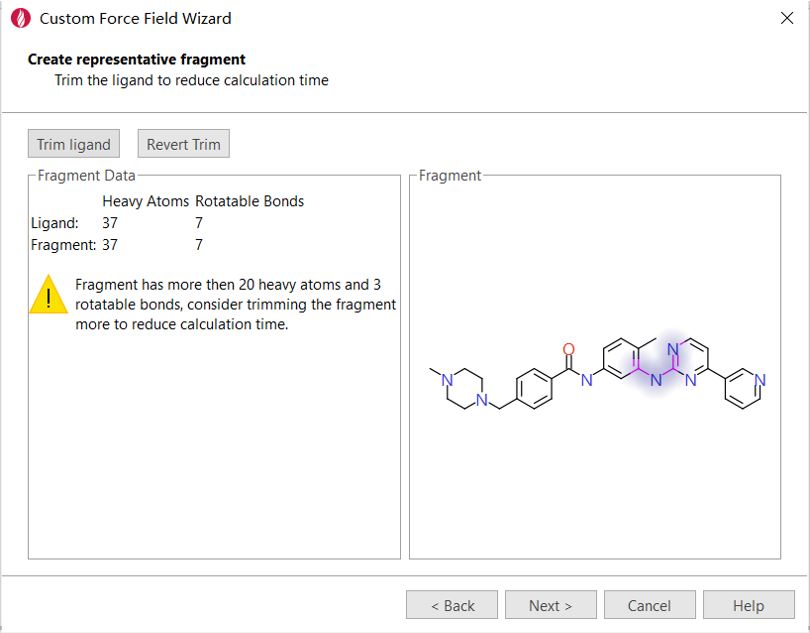
图5. 创建代表性片段
此时,弹出分子编辑对话框,除了图1高亮部分的2-苯氨基嘧啶保留之外,删除其余部分。然后点击Add H按钮,再点击Minimize按钮对2-苯氨基嘧啶进行初步的能量优化。点击OK按钮,退出分子编辑对话框并返回到创建代表性片段对话框(图6)。
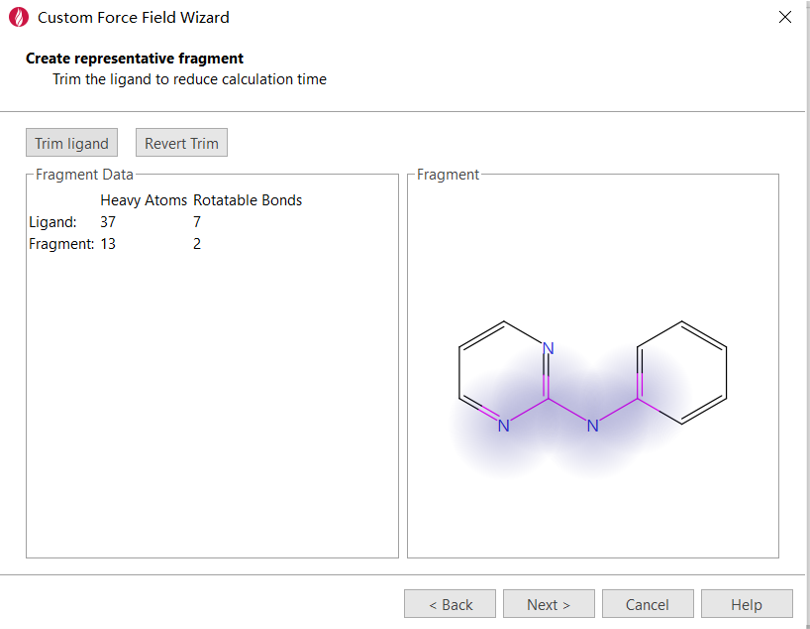
图6. 裁剪后的代表性片段
点击“next”按钮,进入下一步。
3.5 计算参数设置
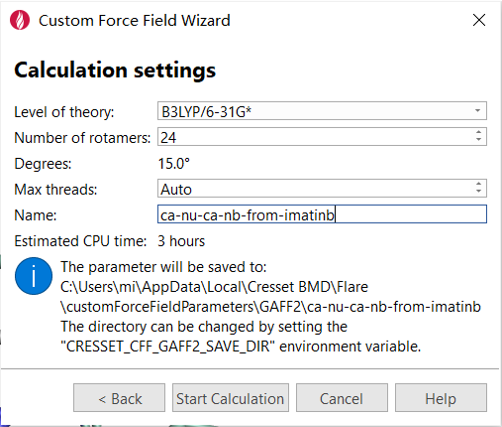
图7. 计算参数设置
Level of theory:理论计算水平。本步骤需要使用QM进行两面角扫描,因此需要指定理论计算采用的方法与基组。Flare/Custom Force Field预设了几种组合, 在Level of theory下来菜单里可直接选择,为了快一点看到结果,可以用B3LYP/6-31G*。
Number of rotomers:柔性键扫描的构象数。默认设置为24,这与下面的Degree步长对应,表示对一个两面角扫描的步长。
Max thread:最大核心数。该值限定了采用多核心进行计算时所用的核心数(每节点)。这里我们用Cloudam云计算,通过broker在云端进行计算,采用每节点16核心进行计算。
Name:给本次计算起个项目名称,也是自定义参数的名称,比如ca-nu-ca-nb-from-imatinb。建议名称可以让你回忆起该计算细节。
点击“Start calculation”按钮开始进行计算。
3.6 用力场参数浏览器分析结果
计算结果可以通过力场参数浏览器(Force Field Parameter Explorer)来分析。点击菜单“Custom Force Field |Show Force Field Parameter Explorer”弹出对话框(图8)。
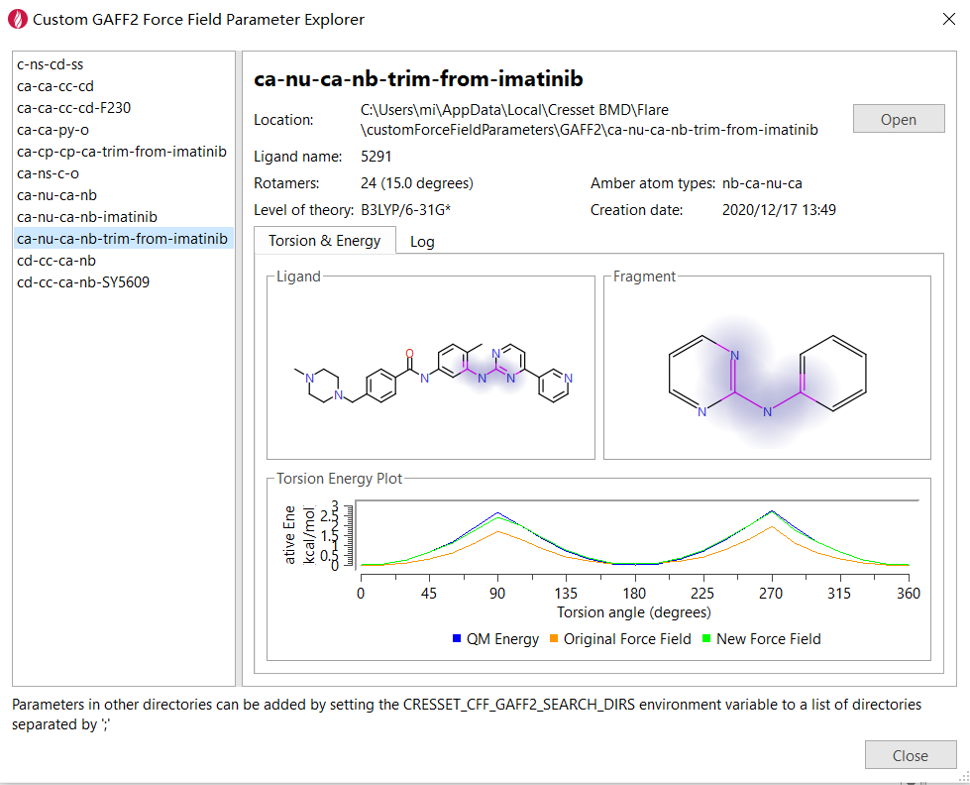
图8. 力场参数浏览器(Force Field Parameter Explorer)对话框
如图8所示,对话框的左边是名称列表,按力场参数名称顺序;右边提供了参数的保存位置、以及QM、原先的力场、优化过后的力场的两面角扫描结果。两面角扫描结果可以方便的判断力场拟合(优化)的效果。两面角扫描的结果,还保存了一份excel格式的文件,点击在Location旁边的Open按钮可以找到Torsionscan.csv的文件,根据该文件绘图得到图9所示的散点图。
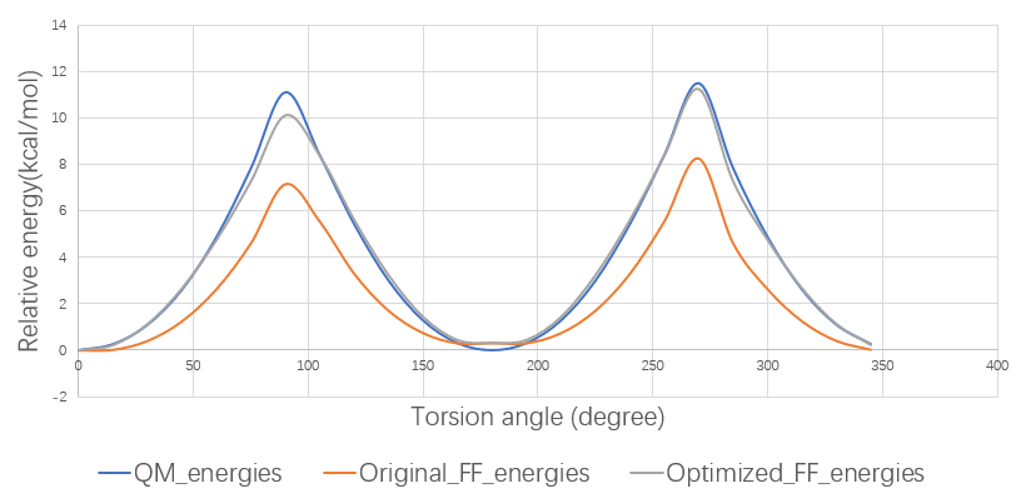
图9. 两面角扫描结果
如图9所示,GAFF2力场(黄色曲线)对2-苯氨基嘧啶描述的并不好,因为黄色曲线与蓝色曲线(基于QM的两面角扫描)很不一致。但是,自定义的力场(灰色)与QM方法拟合的很好,基本重合。因此,可以预见对于该分子,自定义力场会优于默认的GAFF2力场。
用同样的方法可以对Brigatinib进行Torsion profile发现(图10),GAFF2力场(黄色曲线)与QM(蓝色曲线)差异巨大,这说明GAFF2不能良好地描述该化合物(见图10);但是自定义力场(灰色曲线)与QM方法重合地良好。这说明,即使在大部分情况下能很好描述分子的GAFF2力场可能不能正确描述常见的激酶抑制剂片段,因此自定义力场是很有必要的。
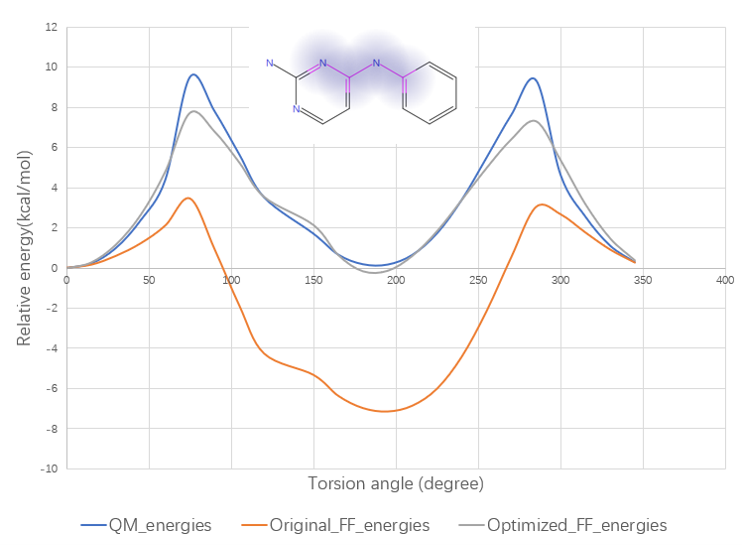
图10. Brigatinib的两面角扫面结果。默认的GAFF2(黄色)不能正确地描述该片段,优化过的力场(灰色曲线)与QM方法(蓝色)拟合地较好。
4 在分子动力学模拟或FEP中使用自定义力场
图11. 分子动力学模拟对话框
Flare的分子动力模拟(Dynamics)与FEP模块支持使用自定义力场以提高模拟的精度。以分子动力学模拟为例(图11),点击对话框“Custom parameters”行的change按钮,弹出力场参数浏览器(见图12)。
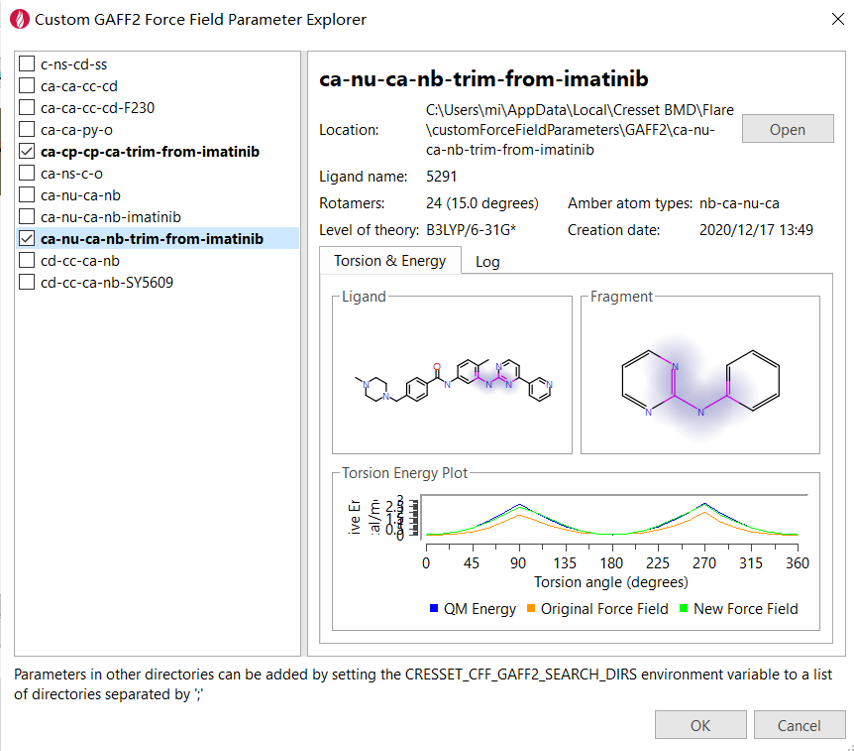
图12. 使用自定义力场参数
在左侧浏览器栏点击感兴趣的力场参数,右边会给出参数的细节;点击自定义力场前的方框,则表示使用该力场参数。选择完力场之后,点击“OK”按钮返回分子动力学模拟的对话框。此时,Custom Parameters行出现数值2,这表示使用了两个自定义力场参数。
现在,点击“Start”(图12),就可以采用自定的力场开始计算了。
Flare/Custom Force Field用于QM-based Torsion profiling
如前所述,Flare/Custom Force Field本质上进行了基于QM的Torsion profile,因此可以用于纯粹的Torsion profile目的,更多的介绍,请阅读:势能面扫描在Torsion Profile中的应用


