
摘要:细胞周期依赖性激酶7(CDK7)由于其在转录和细胞周期进程中的双重作用而具有独特的功能。 CDK7在各种类型的癌症中普遍表达,其下调导致细胞增殖减少,CDK7被认为是可行的癌症治疗靶标。 CDK7抑制剂的开发已经获得了鼓舞人心的进展,两款抑制剂CT7001和SY-1365已经进入临床研究阶段。 本文讨论了CDK7在癌细胞中所起作用的最新理解,并综述了CDK7抑制剂及其在各种癌症模型中的药效以及临床进展。
编译:肖高铿/2020-12-10
原文:Diab, S.; Yu, M.; Wang, S. CDK7 Inhibitors in Cancer Therapy: The Sweet Smell of Success? J. Med. Chem. 2020, 63 (14), 7458–7474. https://doi.org/10.1021/acs.jmedchem.9b01985.
背景
在过去的几十年中,全球范围内轰轰烈烈的抗癌研究一直致力于剖析癌症回路并揭示适合患者个性化疗法(patent-Tailored Therapies)的关键靶标。已有确凿的证据支持干扰许多癌症类型转录程序的遗传变异可作为潜在靶标,并激起了人们对开发阻止过度激活转录疗法的热情。另一方面,细胞周期失调是癌症的标志,靶向细胞周期调控因子的药物已经上市。鉴于细胞周期蛋白依赖性激酶7(CDK 7)在许多癌症的失调转录和细胞周期进程中的作用,CDK7已成为人们广泛追求的抗癌治疗途径[1]。
CDK是丝氨酸/苏氨酸蛋白激酶,可调节转录(CDK 7-13和19-20)或细胞周期进程(CDK 1-6和14-18)[2]。几乎所有的CDK激活都通过:(1)与细胞周期蛋白(Cyclin)结合;(2)T-Loop被CDK激活激酶(CDK Activating Kinasse,CAK)磷酸化。CAK是一种三元复合物,由CDK7、细胞周期蛋白H(Cyclin H)与环-指蛋白MAT1(RING-finger protein MAT1)组成。CDK7的独特之处在于其同时涉及转录与细胞周期的调控。
CDK7的T-loop上的Thr170被自磷酸化并与Cyclin H结合之后,CDK7即被激活。然而这种自磷酸化并不是CAK活性所必须,因为这与CDK7与MAT1的相互作用是匹配的。MAT1是三元复合物所必须的,因为CDK7与Cyclin H的装配并不稳定。与其它CDK仅在T-loop上有一个磷酸化位点不同的是,CDK7有第2个磷酸化位点Ser164,该位点的磷酸化增强其与Cyclin H的结合亲和力。CDK7这两个残基位点的磷酸化均由CDK1与CDK2完成,后两者正反馈调节细胞周期调节因子影响基因表达[4]。
CDK7协调RNA聚合酶II(RNA polymerase II, RNAP II)转录周期的不同段(phase)。在起始阶段,CDK7作为人类一般转录因子II(transcription factor II human,TFIIH)的一部分,RNAP II羧基端结构域(Carboxy-terminal domain,CTD)七肽重复序列的Ser5磷酸化,以促进启动子逃逸(promoter escape)。Nilson等人最近提出CDK7抑制剂可以削弱RNAP II的停顿(paussing)与mRNA封端(capping)作用。THZ1是一种CDK7/12/13的小分子共价抑制剂,它与CDK7的结合避免了DRB-敏感诱导因子(DRB-sensitivity inducing factor,DSIF)与负延伸因子(negative elongation factor,NELE)与RNAP II的结合,从而阻止了转录的生产性延伸。相反,CDK7通过磷酸化CDK9,一种正转录延伸因子b(P-TEFb)的成分,来释放停顿,CDK9继而磷酸化CTD的Ser2残基,从而促进生产性转录8。另一方面,有充分的证据支持相分离(phase separation)在转录调节中的作用[9-15]。各种转录调节子含有低复杂度的无序区,通过瞬时相互作用形成液体状的枢纽(Hub)[9-12]。随后, Hubs可能会发生液-液相分离, 因此以有序的方式划分转录装置。这提示了CDK7在调控基因转录的相分离中起着重要作用[13-15]。RNAP II的CTD是一个低复杂度无序区,聚集在枢纽中。与Boehning等人的研究一起,Lu等人提出RNAP II的未磷酸化CTD通过疏水相互作用进行相分离,而其被CDK7磷酸化后促进其渗入到由细胞周期蛋白T1和组氨酸富集结构域通过静电相互作用形成的相分离液滴中[13,15]。Boehning等人[13]提出这种磷酸化作用使得RNAP II从枢纽中释放出来,从而激活转录延伸。最后,CDK7磷酸化许多转录因子,包括p53和核激素受体,例如雄激素受体(ARs)和雌激素受体(ERs),从而调节其活化和靶基因的表达[16]。Fisher全面综述了CDK7在转录中的作用[1]。
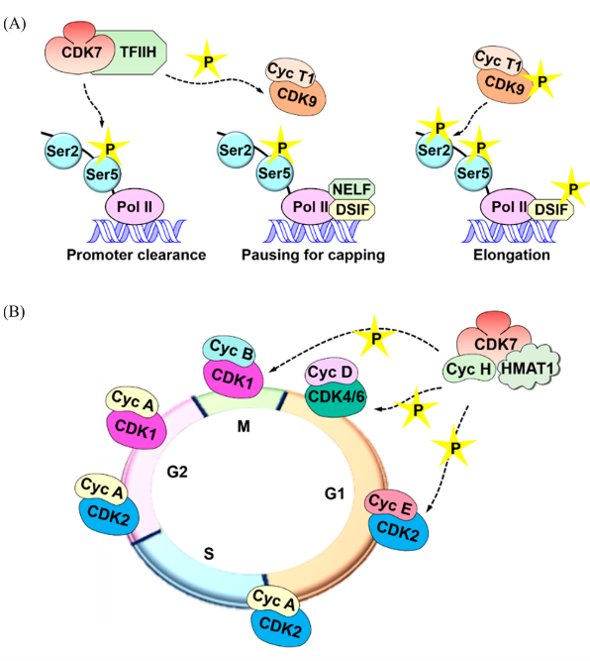
图1. CDK7在细胞转录与细胞周期中的作用
除了其在转录中的作用之外,CDK7通过其CAK功能调节细胞周期进程(图1B)。 已有研究证实CDK7在使CDK4/6磷酸化视网膜母细胞瘤(Rb)蛋白促进G1期进展、CDK2促进G1期向S期进展、CDK1调节G2/M期过渡中的作用[17,18]。CDK7的磷酸化似乎在调节细胞周期中细胞周期蛋白的配对顺序和进程。 例如,CDK7以单体形式使CDK2磷酸化,但与Cyclin H形成复合物影响CDK1的磷酸化。这将使CDK2相对于CDK1在细胞周期中优先与Cyclin A结合。尽管CDK7介导的CDK1/2磷酸化仅对其激活至关重要,但也是CDK4/6激活并保持活性所必须的。在G0-G1进程中会刺激CDK7的磷酸化,从而导致CDK4的激活,并推测在细胞周期的早期阶段还存在一个未知的CDK7激活激酶[18,19]。是否存在CDK7以外的CAK一直也是个争议。先前的研究表明,在T121-表达CDK7mut/mut小鼠胚胎成纤维细胞(MEFs)中,CDK1/2保持磷酸化,这意味着Rb家族蛋白失活后CDK1/2的非CDK7介导的磷酸化(T121是失活Rb家族的多肽)[20]。相反,缺失CDK7的MEF削弱了细胞周期CDKs的磷酸化,并且CDK7抑制剂通常在体外没有CAK活性。因此,尽管CDK7被认为是主要的CAK,但对磷酸化其它CDKs的CDK7非依赖性激酶的存在仍不清楚。
癌细胞中的CDK7:上调还是下调
已在多种癌症类型中检测到异常多的CDK7,并与侵袭性临床病理特征和不良预后相关。CDK7在肝细胞癌、胃癌和结肠直肠癌(CRC)中扩增[4–6]。对173份胃癌标本的免疫组织化学分析显示,CDK 7水平升高,这与肿瘤分级有关7。同样,CDK7蛋白在大部分口腔鳞状细胞癌标本中过表达,与T分期增高、无疾病生存率降低有关,表明其可作为预后生物标志物8。与相邻的正常乳腺组织相比,在癌性乳腺组织中的CDK7蛋白和mRNA水平上调9。令人感兴趣的是,CDK7的表达升高与三阴性乳腺癌(TNBC)患者的临床预后不良,但与ER+乳腺癌患者预后更好有关,这一现象可归因于两种乳腺癌类型的异质性遗传背景[9-10]。
Qiu Hong等人[7]用siRNA法敲低(knockdown) CDK7后,发现降低胃癌细胞的增值并增加G2/M周期细胞的数量;Yu bao等人[11]用CRISPR/Cas9基因编辑技术敲低BT549与MDA-MB-231 TNBC细胞里的CDK7之后,可以降低这些细胞的增殖。同样,Miguel等人[12]发现,用腺病毒/慢病毒载体对MEF细胞进行的CDK7基因沉默会对CDK的T-Loop磷酸化带来损害,并降低E2F驱动基因的转录,从而阻止细胞周期并阻止细胞增殖。但是,几乎没有观察到对全局转录的影响。缺乏CDK7的小鼠胚胎面临早期致死性,但在成年幼鼠中没有明显的表型。 增殖率低的器官(例如大脑)保留了其生理参数;然而,高度增殖的组织(如肠道)保持了高水平的CDK7表达,这表明表达CDK7的干细胞库将随着衰老而耗尽,而无法更新。
长期以来,人们一直认为靶向基因转录是有风险的,因为担心这种策略会比正常细胞缺乏对恶性细胞的选择性。但是,这种观点被逐渐改变。一项早期研究表明,Kin28as(一种工程化的Kin28,酵母CDK7的同源蛋白)可被体积更大的腺嘌呤类似物(1-NA-PP1)选择性抑制,但不会破坏整体转录[13]。抗转录剂可以选择性地关闭依赖异常转录的基因以刺激癌细胞的生长,而不会对管家基因的转录产生不利的影响。 总之,这些研究表明CDK7可作为具有足够治疗窗的有前途的抗癌治疗靶标。
基于结构的CDK7抑制剂设计
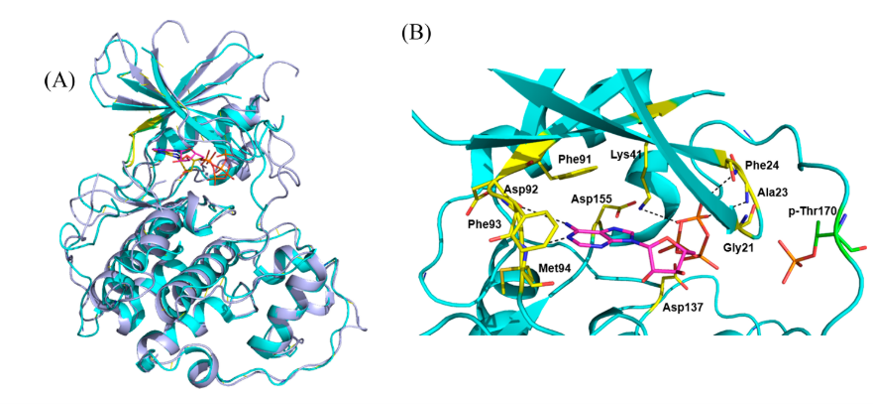
图2. CDK7(PDB 1UA2,青色)与CDK2(PDB:1HCK,淡紫色)晶体结构的叠合
发现CDK抑制剂长期以来一直学术与工业界追求的目标,过去几年已经发现无数的泛CDK抑制剂(pan-CDK inhibitos)。由于缺乏选择性,第一代的CDK抑制剂表现出很强的副作用,这延缓了临床开发。泛CDK抑制剂超出了本文讨论的范围,但已有大规模的综述[2,30,31]。相反,鲜有综述描述CDK7抑制剂[32,33]。这激励了我们撰写本综述,聚焦于最先进的具有优化选择性的CDK7抑制剂。
结构指导的选择性CDK7抑制剂设计主要基于先前的泛-CDK抑制剂,但也基于该激酶的唯一晶体结构(PDB ID:1UA2)[34]。后者表明,CDK7有一个公共的双叶支架出口的ATP结合位点,由 N端(残基13−90)的β-片层以及α-螺旋与C端(残基97−311)α-螺旋组成的缝隙之中。CDK7与CDK2有42%的序列一致性,结构也相似:262个等同Cα原子的RMSD为1.49 Å(图2 A);已有文献综述比较过不同CDK结构的差别[35,36]。ATP的腺苷与CDK7的铰链形成了两个氢键,而三个γ-磷酸氧与Phe23, Ala24, Lys41(图2B)发生氢键相互作用。该晶体结构为开发基于对接和分子动力学(MD)模拟的选择性CDK7抑制剂铺平了道路。
嘌呤类等排体衍生物
为了识别强效、选择性的CDK7抑制剂,Ali等人用Roscovitine作为起点进行基于结构的模拟设计[37]。Roscovitine是第一代泛CDK抑制剂,其结构已广泛用于单一CDK的抑制剂的修饰开发。包括用一系列生物等排体对嘌呤进行替换,比如吡唑并[1,5-a]嘧啶(例如BS-181和CT7001)[37-39]和吡唑并[1,5-a] [1,3,5]三嗪(例如LDC3140和LDC4297)[40]以及不同位置上多样化取代基取代(图3)。基于计算研究,有人提出:以吡唑并[1,5-a]嘧啶或吡唑并[1,5-a] [1,3,5]三嗪骨架替换Roscivitine的嘌呤母核会得到更有效的CDK2抑制剂[41]。吡唑并[1,5-a]嘧啶似乎增强了与铰链区Leu83的相互作用并使得分子的几何形状更加紧凑,从而使其构象更稳定。吡唑并[1,5-a]嘧啶与吡唑并[1,5-a] [1,3,5]三嗪母核之间的结合差异很小。 从一方面看,位置a的氮原子(图3B)防止了该位置的氢原子与相邻的苄基氨基团之间的碰撞,从而稳定了分子的几何。另一方面,氮原子的存在,当与吡唑并[1,5-a]嘧啶偶联时,使得苄基环朝向蛋白质而导致与蛋白质的相互作用降低。 总体而言,两个母核都与CDK2良好容忍,并可进一步用于设计其他CDK抑制剂的合适框架。
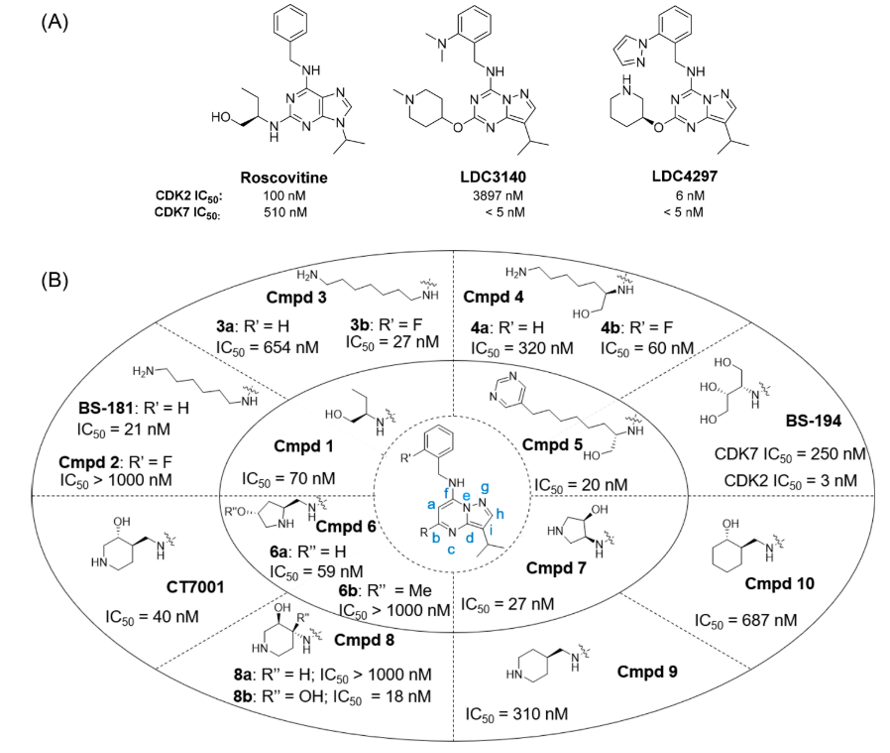
图3. roscovitine、LDC3140与LDC4297的化合物结构(A)以及CDK7抑制剂的结构优化
Ali等人首先根据溶剂化自由能的计算,用吡唑并[1,5-a]嘧啶取代嘌呤,得到化合物1(图3B);吡唑并[1,5-a]嘧啶母核具有较低的、有利的溶剂化能,这有利于其容纳于疏水结合口袋中。比之Roscovitine,化合物1的对接分数稍好,但其与CDK7的结合可进一步改善(IC50 = 70 nM)[42,43]。因此,切除化合物1的羟乙基部分并将吡唑并-[1,5-a]嘧啶基-C5(即图3B中的R位置)处剩余的丙基氨基用1,6-二氨基己基取代(即BS-181)[37]。结构优化主要研究了R位置的各种官能团取代的效应以及苯环(主要是邻氟基)上的取代基对CDK7抑制的影响[42]。根据相应专利中提供的数据,在这两个位点的取代基之间的相互作用决定了CDK7抑制能力。例如,将邻氟基团引入到BS-181的苯环上,即化合物2,急剧降低了CDK7抑制活性(IC50大于1000nM)。相反,在R位置存在1,7-二氨基庚基的情况下,邻-氟取代使CDK7抑制作用增加>24倍(3a对3b)。在(R)-2,7-二氨基庚烷-1-醇而不是1,6-二氨基己基取代基的情况下也是如此,因为通过将邻氟基团引入苯环,CDK7的抑制作用提高了5倍(4a与4b)。同样,在某些情况下,末端带有杂芳基环的分叉侧链的化合物(例如吡啶或嘧啶)仍具有CDK7抑制活性(5,IC50 = 20 nM); CDK7的抑制和选择性取决于侧链的长度和分枝以及其嵌入环的类型。另一方面,较短的羟基化侧链被用于开发CDK2抑制剂,例如BS-194[44]。在整个系列中,BS-181是研究最多的CDK7抑制剂[37]。体内研究表明,经腹膜内注射后,小鼠体内半衰期(t1/2)为405分钟。BS-181不是ATP结合盒(ABC)药物转运蛋白(ATP-binding cassette drug transporter, ABC drug transporter)的底物,但由于其伯胺(pKa = 10.2)在生理pH下会质子化,因此具有较低的生物利用度,从而降低了该分子的胃通透性[45]。
BS-181结构优化的主要目的是提高其口服生物利用度[38]。主要对R-位用氨基哌啶、氨基甲基哌啶、氨基吡咯烷和氨基甲基吡咯烷进行取代修饰。CDK7抑制随着取代基性质(包括立体化学)变化而变化。比如6a的羟基被甲基掩盖之后,CDK7抑制活性急剧下降(6a vs 6b)。R位的(3R,4S)-4-氨基吡咯烷-3-醇,(3S,4S)-4-Aminomethylpiperidin-3-ol,取代后仅得到一个强效CDK7抑制剂(7, IC50 = 27 nM),但是其对CDK1与CDK2的选择性也分别达到48与146倍。(3R,4S)-4-氨基吡咯烷-3-醇耐受(IC50 = 40 nM)良好,并由此发现了CT7001 (ICEC0942),对CDK1与CDK2的选择性分别达到了37与14倍。CT7001的差向异构体(8a)对CDK7的活性很差,IC50大于1000nM。有趣的是,在8a的哌啶环C4处引入一个羟基(即8b),不仅恢复了CDK7的抑制作用(IC50=18nm),而且对CDK1(77倍)和CDK2(113倍)表现出优异的选择性。去掉CT7001的羟基或者将哌啶环用环己烷替换(比如化合物9、10)会导致对CDK7的抑制活性分别降低7、17倍。CT7001在进一步开发中,它的口服生物利用度为30%[38-39]。另外,CT7001的外排率(efflux ratio)高达86.5%,血浆蛋白结合率为90.8%,CYP3A4的IC50=5.1uM, 小鼠t1/2=1.9hr。同时CT7001为ABC家族B1 (ABC subfamily B member 1, ABCB1)的底物,这是一种转运蛋白,它介导了药物耐受[46]。CT7001的合成路线如图S1所示。
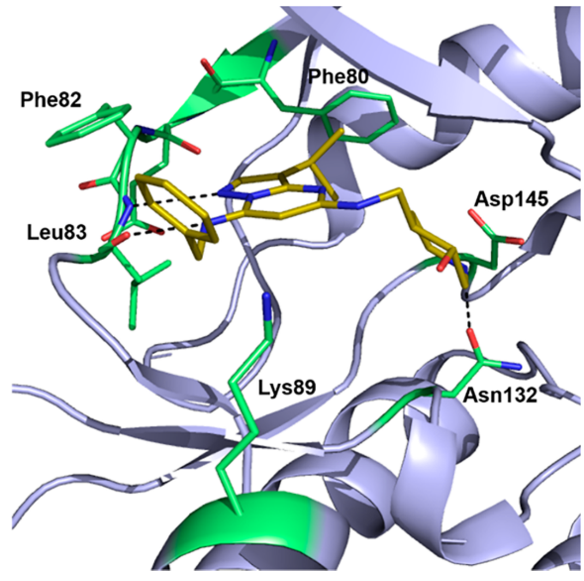
图4. CT7001(黄色)与CDK2(浅紫色)的共晶结构(PDB ID: 5JQ5)
各种获得CT7001与CDK7共晶体结构的努力均未成功。因此,Hazel等人解释了CDK2与CT7001(PDB ID:5JQ5)复合物晶体结构[42]。该共晶体结构表明,吡唑[1,5-a]嘧啶母核在铰链区与Leu83主链形成两个氢键相互作用。 第三个氢键在哌啶环的氮和Asn132之间形成;前一个片段还与Asp145有静电接触(图4)。
MD模拟和等温滴定量热法(ITC)被用于推测CT7001与CDK7的结合模式,并用于理性设计对CDK7/CDK2选择性抑制剂[47]。序列比对表明,CDK2和CDK7的ATP结合位点的许多残基是保守的,作者认为结合能力的差异可能与结合位点的局部结构有关,并受两种蛋白质固有的柔性影响。使用CDK2-CT7001(PDB ID:5JQ5)共晶体结构在CDK7(PDB ID:1U2A)的晶体结构中产生CT7001的起始结合模式,并用于MD模拟。CT7001在CDK7中采用了与CDK2相同的结合方式,并保持与铰链区,特别是CDK7-Met94的氢键相互作用。此外,它能够与CDK7的Asp137形成额外的相互作用,这在CDK2的情况下是不可能达到的。最重要的是,CT7001可以通过极性相互作用桥接CDK7的C端Asp155和富含甘氨酸loop的Gly21,从而限制了loop的柔性。然而,在使用CDK2进行的模拟中,仅5%检测到这种相互作用,这可能是CT7001与CDK2的相互作用力弱于与CDK7相互作用力的原因。实际上,用ITC法分析CT7001与重组CDK7Asp155Ala和CDK2Asp145Ala的结合亲和力实验结果表明,CT7001对CDK2Asp145Ala
保持着与野生型CDK2相同的结合亲和力,而其对CDK7Asp155Ala的结合比对野生型CDK7弱3-4倍。同样,CDK7中的Val100使得其比相应的CDK2-Lys89口袋更大。ITC研究显示,CT7001与CDK7Val100Lys的结合不如与野生型CDK7来的强,这证实了CDK7中结合位点的大小对CT7001的激酶选择性也有作用。
对BS-181为中心化合物库的筛选以及对此类苗头化合物的药物化学优化发现了化合物LDC3140和LDC4297(图3)[40,48]。两者都含有吡唑[1,5-a][1,3,5]三嗪为母核,哌啶酰氧基取代了吡唑[1,5-a]嘧啶C5处的氨基链接臂(linker)以及取代的N-苄基。该骨架的衍生物被认为是有效的CDK7的ATP-竞争性抑制剂。两个LDC化合物在一组150种激酶中均表现出CDK7的良好选择性。
共价抑制剂
THZ1是首个共价CDK抑制剂(图5)。它与位于ATP结合位点外面的半胱氨酸结合,因此为提高对CDK7的选择性提供了无与伦比的机会[49]。THZ1包含N-苯基-嘧啶-2-胺骨架,该骨架是许多激酶抑制剂的公共母核,包括伊马替尼和CDKI-73(最有效的CDK9抑制剂之一)[50]。独特之处在于,THZ1包含了一个丙烯酰胺片段,负责与附近Cys312不可逆结合的弹头。实际上,没有这种弹头的THZ1-R对CDK7和细胞增殖的抑制作用较小。而且,CDK7Cys312Ser不受THZ1抑制,进一步证明与Cys312的共价键具有最佳抑制作用。与CDK7相似位置具有半胱氨酸的CDK12/13也被THZ1抑制。该化合物在各种小鼠模型中均能显着抑制肿瘤生长(表1);但是,它是ABC药物转运蛋白的底物,引起了人们对潜在耐药性的担忧[46,51]。
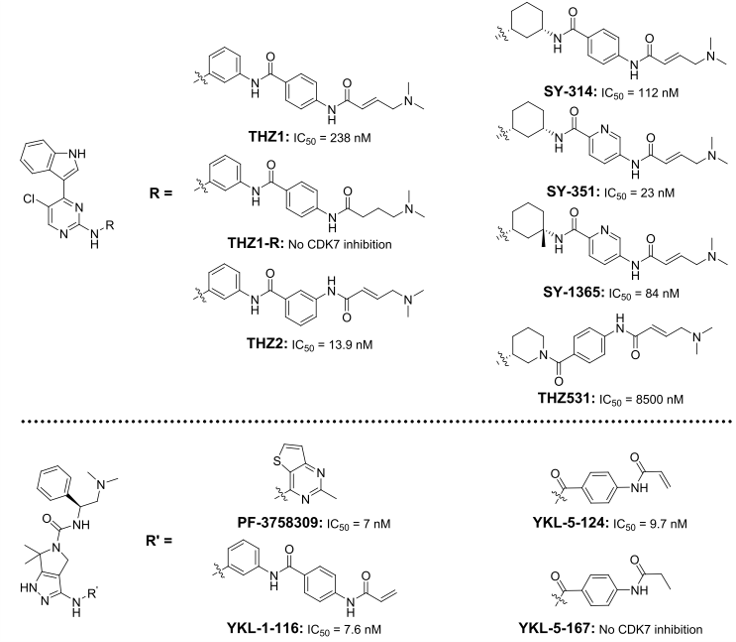
图5. CDK7共价抑制剂化学结构
对THZ1进行结构优化迭代以提高其体内代谢稳定性(小鼠血浆中t1/2 = 45min,CL = 129 mL/min/kg)[28,52]。将丙烯酰胺从苯甲酰胺的对位移到间位(THZ2,图5),其体内t1/2延长了5倍,同时保留了对CDK7的选择性抑制。用1S,3R-环己二胺取代间苯二胺(即SY-314)会使小鼠的清除率降低7.6倍(CL = 17 mL/min/kg),但这降低了共价抑制的效果。用吸电子的吡咯酰胺取代苯甲酰胺,即SY-351,恢复了细胞靶标结合,但清除率增加到29 mL/min/kg。迭代优化涉及在环己二胺片段上引入甲基以获得SY-1365。SY-1365通过增加酰胺链接臂周围的立体障碍以限制弹头的可及构象。SY-1365的清除率明显降低,为5.6 mL/min/kg,更高的Kinact/KI值为0.131μM-1s-1。值得注意的是,用哌啶取代环己胺得到了THZ531,其对CDK12的选择性是CDK7的53倍(IC50 158 nM对8,500 nM)[51,53]。方案S2中描述了合成SY-1365的合成路线。
除CDK7外,THZ1对CDK12/13的抑制作用模糊了CDK7抑制作用的细胞表型[54];这促使人们寻求更具选择性的CDK7抑制剂。PF-3758309(图5)是一种对CDK7具有抑制作用的PAK4抑制剂[55-57],将THZ1的共价弹头与PF-3758309的吡咯烷基吡唑骨架杂交产生了YKL-1-116 。后者保留了对CDK7的抑制活性和选择性(IC50 = 7.6 nM),但在体外实验中显示出较低的抗增殖活性[57]。为了提高活性,结构修饰考虑了优化共价弹头的长度和轨迹。去掉苯胺以缩短了共价弹头,得到YKL-5-124。它保持了通过丙烯酰胺部分共价结合的能力,因此保留了对CDK7的亲和力(IC50 = 9.7 nM)[54]。实际上,YKL-5-124是一种更强效的抑制剂,比之THZ1,对CDK7具有更快的动力学速度(Kinact/ KI = 103 nM-1μs-1与9 nM-1μs-1);在测试浓度(10-4至10 mM)下,它也没有表现出对CDK12/13的抑制作用,并且对CDK7的选择性是CDK2/9的130倍以上(IC50分别为1300和3020 nM)。 YKL-5-167(其中的丙烯酰胺被丙酰胺替代)在体外激酶活性测定中失去了抑制CDK7的能力。
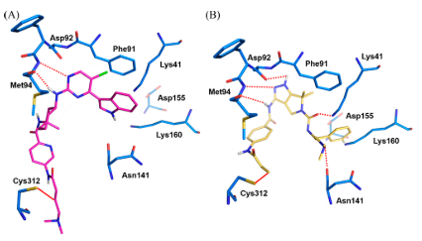
图6.预测的SY-1365(A)以及YKL-124与CDK7的结合模式
对接研究可预测上述共价抑制剂与CDK7的结合模式[49、52、54],同时也可用来指导进一步的结构设计。THZ1和SY-1365在CDK7结构模型中采取了相似的结合模式。除与Cys312形成共价键外,嘧啶-2-胺的N1和C2-NH还通过两个氢键与Met94的主链结合,氯与gatekeeper Phe91形成疏水相互作用(图6A)。就SY-1365而言,1,3-环己烷二胺母核改善了形状互补性和疏水性,与THZ1相比,具有更高的活性和选择性。另一方面,YKL-5-124通过其3-氨基吡唑并吡咯烷母核与铰链区相互作用,与Asp92和Met94形成了三个氢键相互作用(图6B)。 此外,还与Lys41、Asn141形成了两个氢键相互作用,这增强其对CDK7的结合亲和力。
在研CDK7抑制剂
表1. 在研CDK7抑制剂
| Structure | Code | CDK Inhibition, IC50(nM) |
|
|
BS-181 |
CDK1 8100CDK2 880CDK4 33000CDK5 3000CDK6 47000CDK7 21CDK9 4200 |
|
|
CT7001 |
CDK1 1800CDK2 620CDK4 49000CDK5 9400CDK6 34000CDK7 40CDK9 1200 |
|
|
THZ1 |
CDK7 238CDK12 893CDK13 628 |
|
|
THZ2 |
CDK1 96.9CDK2 222CDK5 134CDK7 13.9CDK8 6830CDK9 194 |
|
|
SY-1365 |
CDK2 2117CDK7 84CDK9 914CDK12 204 |
|
|
YKL-1-116 |
CDK2 1140CDK7 7.6CDK9 大于10000 |
|
|
YKL-5-124 |
CDK2 1300CDK7 9.7CDK9 大于3020 |
|
|
SY-5609 |
CDK2 2900CDK7 0.06*CDK9 970CDK12 770 |
| 结构未公开 | UD-017 |
CDK1 大于10000CDK2 6200/8900CDK3 大于10000CDK4 大于10000CDK5 5100CDK6 大于10000CDK7 16CDK9 大于10000 |
BS-181
现有的CDK7抑制剂已被开发为化学探针,以阐明CDK7在癌细胞生物学中的作用,并在体外和体内研究CDK7抑制是否会转化为有效的抗癌策略。 BS-181是第一种选择性CDK7抑制剂(IC50=21nM);对CDK7的活性力是对CDK2和其他一些CDK1/4−6/9家族成员的41倍以上(表1)。与69种非CDK激酶相比,BS-181在10μM时全部的非CDK激酶剩余活性大于15%[37]。体外实验证实BS-181对MCF-7乳腺癌、BT549和MDA-MB-231 TNBC和各种胃癌细胞系的体外生长具有抑制生长的能力,包括诱导细胞凋亡并抑制细胞周期进程[27,37,58]。它降低了TNBC细胞中RNAP II CTD的Ser5/7磷酸化,提示mRNA转录停滞[27]。与对照组相比,当腹腔注射10和20 mg/kg bid 14天时,BS-181分别使MCF-7乳腺癌异种移植物的生长减少25%和50%,且未检测到毒性[37]。使用相同的方案,与对照组相比,在BGC823胃癌移植瘤模型中,BS-181以剂量依赖的方式抑制肿瘤生长[58]。总而言之,BS-181提供了CDK7抑制剂抗乳腺癌和胃癌效果的首个概念验证,并提示靶向转录是有效的治疗方法。然而,BS-181口服生物利用度低,阻碍了其进一步的发展。
CT7001
CT7001是BS-181口服有效的衍生物。它保留了与BS-181类似的CDK7抑制活性,并具有对CDK1/2,4-6/7/9的选择性(它们的IC50高了1-3个数量级,表1)和对许多非CDK激酶也具选择性[39]。当针对一组60种癌细胞系进行评估时,中位GI50=0.25μM,并导致一组患者来源的小细胞肺癌(SCLC)细胞系死亡。CT7001激活Caspase 3/7并促进PARP裂解,这意味着细胞凋亡,HCT-116细胞在暴露24小时后其细胞周期停滞在G2/M期。HCT116 CRC和MCF-7乳腺癌异种移植物的小鼠分别以100 mg/kg/day的剂量口服给药时,CT7001分别在第13天和第14天将肿瘤生长抑制60%(p = 0.0001)。CT7001还用TNBC的小鼠模型进行了评估,可导致肿瘤强而持续地消退,并且对体重的影响很小(小于10%)[59,60]。小鼠每天一次口服CT7001可致MV-4-11急性髓细胞白血病(AML)异种移植模型完全转归(消失)[61]。在28天的毒性研究中,CT7001的耐受性也很好,使用的大鼠和狗没有中性粒细胞减少的迹象[59]。该化合物目前处于I期临床试验(NCT03363893),适应症为三阴性乳腺癌与前列腺癌,实验预期与2021年3月份完成。
THZ1与THZ2
作为首个CDK7共价抑制剂,THZ1是靶向CDK的不可逆抑制剂的重大突破[49,62]。THZ1抑制CDK7的IC50为238 nM,但仍抑制CDK12/13(IC50分别为893和628 nM)[54]。其抗增殖能力与BS-181相似,对一组含598种癌细胞系的IC50值小于200 nM,这一作用主要归因于其调节(原癌基因)转录因子的表达[49]。T细胞急性淋巴细胞白血病(T-ALL)Jurkat细胞暴露于THZ1会抑制细胞增殖并伴随着MCL1和XIAP等抗凋亡蛋白的表达,从而导致细胞凋亡。它还减少了CDK1/2的磷酸化,从而导致细胞周期停滞在G2。用高剂量THZ1处理Jurkat细胞会降低总体mRNA水平,而低剂量的暴露会下调一个基因子集,包括与超增强子(SE)相关的基因RUNX1。THZ1对源自患者的T-ALL与慢性淋巴细胞性白血病细胞的有效,在T-ALL异种移植小鼠模型中没有观察到全身毒性。
THZ1的发现推动了评估CDK7抑制剂对侵袭性肿瘤的潜在价值的努力,对于侵袭性肿瘤目前存在未满足的临床需要。 早期研究发现,THZ1对SE驱动基因优势表达的癌症特别有效[49,65,67,68]。虽然转录依赖于众多调节因子,但SE利用转录机制来促进其靶基因的高水平表达;因此,靶向转录调控因子(包括CDK7)有可能损害SEs促进的转录。用THZ1处理异种移植了小细胞肺癌的小鼠产生了明显的肿瘤应答,这是由于扩增的C-MYC和MYCN原癌基因易受THZ1的影响[65]。当用THZ1处理黑色素瘤细胞系时,这一概念得到了反映,确定了SE相关的MITF和SOX10在人类黑色素瘤亚群中容易受到THZ1的影响[67]。THZ1通过在MITF和SOX10处拆卸SE装置并阻止其致癌转录来抑制体外黑素瘤细胞的生长和在体内抑制异种移植肿瘤的生长。 在神经母细胞瘤中,与没有该基因扩增的细胞相比,MYCN扩增的细胞对THZ1更敏感10倍, 这似乎是药物下调了MYCN和SE相关基因转录的结果[68]。 THZ1诱导高表达MYCN的细胞凋亡并在体外停止其细胞周期,在小鼠异种移植模型中显示出体内疗效,同时没有明显的毒性, 这进一步突出了抗转录剂在癌细胞中下调SE相关基因中的选择性,而对正常细胞中的整体转录几乎没有影响。
此外,将携带STAT3Tyr640Phe(一种具有高转录活性的突变)的OCI-Ly12外周T细胞淋巴瘤(PTCL-NOS)细胞与THZ1一起温育,下调了高转录STAT3靶基因(包括MYC、PIM1和MCL1)的表达[89]。THZ1触发与caspases 3/7激活以及PARP裂解有关的细胞死亡,并抑制RNA P II CTD的Ser2磷酸化,但在暴露9小时后对细胞周期的干扰最小。这表明CDK7转录活性是PTCL-NOS细胞生存所必需的。
三阴性乳腺癌(TNBC)因其具有侵润性而广为人知[27],THZ2可挑战TNBC。THZ2是THZ1的间位异构体,但对CDK7的抑制活性高一个数量级(表1)。 Wang等人认为,TNBC的生长依赖于编码转录调节因子和信号转导成分的基因汇编[28]。THZ2提供了靶向这种“Achilles cluster”基因的机会,而不仅仅是像其他“靶向疗法”那样是一些致癌驱动因子。值得注意的是,TNBC中“Achilles cluster”的基因中只有40%与SE相关。据推测,SE非相关基因依赖于SE驱动的转录因子的持续表达,因此是次级应答者。与此相一致,Greenall等人的研究表明,高级别胶质瘤对THZ1的敏感性与线粒体核糖体蛋白(MRP)家族的下调有关,而线粒体核糖体蛋白(MRP)家族也与SEs无关,这表明可能还有其他因素有助于抑制剂的疗效[90]。THZ1下调了癌基因的受体酪氨酸激酶(RTK)家族,包括EGFR、PDGFR、MET、AXL及其下游效应子,即AKT、ERK和STAT3通路,并通过将细胞阻滞在G2期引起DNA损伤。
另一方面,THZ1对耐药的癌症也显示有效。例如,它在体外对乳腺癌T47D palbociclib耐药(PDR)细胞系具有显著的生长抑制作用[91]。此外,用THZ1治疗后在体外还恢复了对enzalutamide(苯扎鲁胺)耐药的去势耐药前列腺癌(CRPC)细胞的生长抑制,并且在AR-阳性前列腺癌的小鼠中导致显著的肿瘤生长抑制[72]。患有CRPC的患者由于AR信号的恢复而对雄激素剥夺疗法(包括enzalutamide)产生抗性。研究表明,CDK7抑制阻止了RNA聚合酶II转录亚基1(MED1)介体的磷酸化,从而阻断AR信号传导和配体激活的MED1向染色质的募集。尽管承认有必要进行进一步的相分离研究,Rasool等人提出了CDK7介导的MED1磷酸化在MED1缩合物形成中的作用,MED1缩合物触发了装置的组装并激活了SE驱动的转录。
THZ1或THZ2对肝细胞癌[22,26]、胃癌[21]、宫颈癌[70]、胰腺癌[73]、结直肠癌[69]、非小细胞肺癌(NSCL)癌[92]以及SOX-2扩增的肺鳞状细胞癌[93]的疗效进一步得到体内与体外的实验证实。随着诱导细胞凋亡和/或细胞周期进程受损,这些癌症类型的细胞因暴露于THZ1或THZ2而死亡[21-23,66,69,70]。对于肝癌,在以MYC的异位表达为特征的细胞类型中观察到对THz1的更高敏感性[22]。此外,THZ1显示出抑制NSCL癌细胞中的糖酵解并显着抑制其迁移[92]。实际上,THZ1在细胞迁移中的作用仍存在争议。在一项研究中,THZ1增加CRC细胞的迁移性并通过蛋白激酶D1(PKD1)/snail通路触发它们的转移[23]。然而,其他研究表明,在伴有淋巴结转移的食管鳞状细胞癌中和在乳腺癌中,CDK7/cyclin H与C-末端结合蛋白2(CtBP2)的相互作用促进了细胞侵袭,而CDK7或cyclin H的沉默导致CtBP2的泛素化,从而阻碍了上皮-间质的转化[94,95]。似乎需要激活CDK7而不是其它的激酶以激活CDK7/cyclin H-CtBP2轴并增加细胞迁移。与此相一致的是,敲低BT549和MDA-MB-231 TNBC细胞中的CDK7可以减少细胞迁移[27]。除此之外,还发现THZ1抑制了肌源性分化,这引起了人们对肌肉功能潜在副作用的担忧[96]。总的来说,THZ1/2在体外和体内对各种顽固性肿瘤有效,没有明显的全身毒性。 尽管两者都没有超过临床前研究的进展,但都为发现SY-1365(第一个已经进入临床试验的CDK7抑制剂)奠定了基础。
SY-1365
对THZ1的迭代结构优化产生了SY-1365,其具有更高的药效(表1)、更高的共价抑制CDK7效率以及增强的稳定性(kinact/KI=0.131 μM−1s−1vs 0.003 μM−1s−1;CL=5.6 mL/min/kg vs 129 mL/min)[52]。在表观Km的ATP浓度下,SY-1365对CDK7的IC50为84 nM,并且对CDK12仍然保留有抑制作用(IC50 = 204 nM)。SY-1365对比CDK2与CDK9的选择性分别为25倍和10倍(表1)。SY-1365通过下调致癌转录因子、抗凋亡蛋白(例如,MCL1和MYC)、调控细胞周期与DNA修复(尤其是,同源重组修复和错配修复)进展的通路来介导其抗癌活性,从而导致THP1细胞的凋亡[52]。值得注意的是,尽管靶标被完全占据,但在hTERT永生化的非癌细胞中观察到的凋亡却很小。在多种体内小鼠模型中评估了SY-1365的药效,包括AML(Kasumi-1和ML-2),卵巢(OVCAR3和患者来源的异种移植(PDX)OV15398)和TNBC(HCC70,MDA-MB-468,BR1458 PDX和BR1282 PDX),每周一次或两次静脉注射30−40 mg/kg时,会导致肿瘤消退[52,76−79,97]。SY-1365耐受性良好,没有骨髓抑制的迹象且极少体重减轻。2017年5月,开始了一项分为两部分的I期临床研究。第1部分评估了逐步增加剂量的SY-1365静脉注射对经重度预治疗的晚期实体瘤患者的安全性,第2部分评估对单一用药或联合用药治疗对卵巢癌和乳腺癌的疗效(NCT03134638) [81]。最初的临床数据显示,有87.5%的患者退出治疗,主要是由于疾病的进展。剂量限制毒性为轻度事件,例如头痛和疲劳。未检测到嗜中性白血球减少症。随后在总共19名可评价反应的患者中报告了初步临床结果 [80]。
YKL-5-124
THZ1作为选择性CDK7抑制剂的成功并未被视为神圣不可侵犯,因为其作用机理与选择性沉默CDK7的机理不匹配,特别是关于CDK7抑制对转录的影响方面。例如,体积大的ATP衍生物(如1-NMPP1)可以抑制CDK7Phe91Gly/Asp92Glu突变的HCT116细胞,不会像其他癌细胞中的THZ1一样抑制RNAP II CTD的Ser残基的磷酸化[17,49,57]。同样,CDK7基因失活降低了细胞增殖并阻止细胞周期进程,但对转录几乎没有影响 [20]。这促使人们进行研究并发现了更具选择性的CDK7抑制剂YKL-5-124,它不抑制CDK12/13,并已用于破译选择性CDK7抑制在癌症中的作用 [54]。YKL-5-124不能在CDK7Cys312Ser突变的细胞中发挥其作用,这证明CDK7激酶活性的不可逆抑制是YKL-5-124的作用机制。
癌细胞对YKL-5-124的生物学应答与对THZ1和SY-1365的生物学应答不匹配。但是,它复制了CDK7沉默的表型。例如,YKL-5-124抑制CDK1和CDK2磷酸化,导致HAP1和Jurkat细胞中G1和G2/M细胞含量增加,S期细胞含量降低[54]。但是,将这些细胞暴露于YKL-5-124时,PARP未被切割,也没诱导凋亡。这与CDK7/12/13抑制剂THZ1和SY-1365不一致,后者可诱导各种类型的癌症发生细胞凋亡[49,52,68]。另外,即使在高浓度也YKL-5-124也不能抑制Jurkat细胞的RNAP II CTD上Ser5的磷酸化;而与THZ1在低浓度时即可做到[49,52,98]。与THZ1相似,SY-1365在AML(THP1)、TNBC(HCC70)和永生化的正常RPE-hTERT细胞系中以剂量和时间依赖性方式抑制RNAP II CTD的Ser2、Ser5和Ser7的磷酸化[52]。剖析YKL-5-124对E2F驱动基因的影响表明其对细胞周期转录程序具有强效的抑制作用,这与以前的结果一致,证明CDK7的基因失活除了对E2F调控基因外,对全局RNAP II介导的转录没有影响[20]。随后的研究表明,YKL-5-124对细胞增殖的作用主要是由于其以CDK7依赖性方式损害细胞周期进展。相反,THZ1和SY-1365的强大细胞毒性作用是由于抑制RNAP II CTD的Ser残基磷酸化以及基因表达的广泛丧失,这是由于同时抑制CDK7/12/13引起的。实际上,将YKL-5-124与选择性CDK12/13抑制剂THZ531联合使用,可以囊括THZ1和SY-1365对CDK7/12/13更广泛的抑制作用、转录因子基因的表达以及CTD磷酸化[54]。这些发现支持抗癌策略侧重点从针对个体基因改变的范式到攻击供给癌细胞增殖的多个致癌检查点的转移。
CDK7抑制剂与联合用药
与单独抑制CDK7相比,联合用药提高了抗癌活性。YKL-1-116(表1)和直接的p53稳定剂(或5-氟尿嘧啶作为抗代谢药)联用对CRC细胞具有协同的细胞毒性作用[57]。SY-1365与氟维司群(一种选择性的ER调节剂)联用处理乳腺癌 T47D PDR细胞,在较低浓度下表现出协同活性[91]。此外,SY-1365与卡铂联用在卵巢癌移植瘤模型中具有协同作用[99]。与单独使用任何一种药物相比,CT7001(50 mg/kg/天)与他莫昔芬(100μg/天)的联用获得了更大的协同作用,对MCF-7移植瘤模型对肿瘤生长的抑制作用(p = 0.0002),单用他莫昔芬或CT7001的抑制作用p值分别为0.008和0.003[39]。
当与BCL-2蛋白家族的抑制剂联合使用时,CDK7抑制剂显示出对癌细胞生长的增强抑制作用。在PTCL-NOS细胞中,THZ1与obatoclax(一种BCL-2家族蛋白的泛抑制剂)联用在体外、离体和体内均显示出抗淋巴瘤活性,且毒性没有增加[89]。同样,THZ1与ABT-263(一种BCL-2和BCL-XL抑制剂)协同抑制BT549、HCC1143、HCC1937和Hs578T TNBC细胞系的增殖[27]。相似的,同时用THZ1和ABT-263处理的HuCCT1或HuH28胆管癌细胞系通过下调MCL-1而导致体外协同细胞死亡[71]。SY-1365与venetoclax(一种BCL-2抑制剂)联合使用时还可协同抑制Kasumi-1、ML-2、THP1和KG-1 AML细胞系的生长,在KG-1小鼠模型中,具有更高的肿瘤生长抑制率(87.5%),相比之下,单用SY-1365为62.6%,单用venetoclax为48.4%[72]。
另一方面,THZ1可以挽救各种靶向药物耐药的抗癌活性[100,101]。THZ1与BGJ398(FGFR抑制剂)、厄洛替尼(EGFR抑制剂)或克里佐替尼(ALK抑制剂)的组合通过防止在促进抗性发展的基因处形成活性增强子来防止在相应RT112(FGFR)、PC9(EGFR)和H3122(ALK)细胞群体中出现对酪氨酸激酶抑制有抗性的克隆[100]。此外,将THZ1与(i)BGJ39联用处理FGFR突变膀胱癌移植瘤模型或(ii)厄洛替尼联用处理EGFR突变携带NSCLC的小鼠模型,与单独用药相比,联合用药延缓了肿瘤生长。同样,在HCC1569和HCC1954移植瘤模型中,THZ1与lapatinib(一种EGFR和HER2的双重抑制剂)联用可使对HER2抑制剂有抗性的HER2+乳腺癌细胞致敏,并导致最佳且持久的肿瘤消退[101]。同样,对CDK7和BRD4的双重抑制也导致K562 AML细胞株在体外的协同抑制作用,在含有溴-和末端外域(BET)耐药细胞的异种移植小鼠中显示出协同抑制作用[102]。
联合用药也可能使CDK7抑制剂避免了潜在耐药性。最近的研究表明,BRD4的耗尽会使头颈部鳞状细胞癌(HNSCC)细胞对THZ1敏感[103]。THZ1与BRD4抑制剂JQ1联用可通过诱导凋亡和衰老来协同抑制HNSCC细胞的增殖。这种诱导是由BRD4和CDK7介导的SE驱动YAP1破坏引起的。另一方面,MYC扩增的癌细胞系,包括神经母细胞瘤,SCLC和TNBC,对THZ1高度敏感[28,65,68,104]。然而,Lu等人表明,用亚致死浓度的THZ1对胰腺癌细胞的长期治疗,通过下调MYC而导致细胞对药物的获得性抗性[73]。THZ1与脂质纳米颗粒(LNP)配制的靶向MyC mRNA的Dicer底物siRNA(DsiRNA)联用,在肝癌小鼠模型中显示出加和的抗肿瘤功效[105]。然而,对MYC的双重抑制是否会绕过MYC介导的耐药性尚待回答。尽管联合用药似乎可以提供累加效应,但CDK7的选择性抑制在某些类型的癌症中仍然有价值,例如,那些依赖于E2F驱动基因的细胞周期进程。
其它在管线中的CDK7抑制剂
临床前数据已经突显了CDK7抑制剂作为抗癌药的应用潜力,也预示了在临床应用上将有光明的治疗应用前景,这促使人们开发全新的、选择性的CDK7抑制剂。一些新的化学实体也被开发出来处于临床前研究阶段。SY-5609 (Kd = 0.059 nM)是为了克服SY-1365口服生物利用度低而开发[83]。它是一个强效、可口服的CDK7抑制剂,对CDK2/9/12的选择性高达13000-49000倍或更高。在1μM浓度下针对485个激酶的筛选分析表明,仅有9个激酶的抑制率达到了70%以上,其中包括CDK13/16-18。它抑制一系列卵巢癌与TNBC细胞的生长(EC50在6-17nM之间)、阻滞细胞周期并触发凋亡,但不影响非肿瘤细胞。SY-5609口服给药(2.5mg/kg与5mg/kg,每天两次)21天,在TNBC(HCC70)与卵巢癌(OVCAR
3)细胞移植瘤模型上观察到完全的肿瘤消退,肿瘤消退一直持续到最后一次给药后14天。进一步的研究证实了在低于最大耐受剂量(maximum tolerated dose ,MTD) 口服给药( 3-5mg/kg,每天两次)时都能实质性地抑制这两种移植瘤模型的肿瘤生长。MYC于MCL1的下调可作为CDK7地体内抑制活性的标记物。临床前研究支持其当前进展足以通过IND申请,以期于2020年启动I期试验。
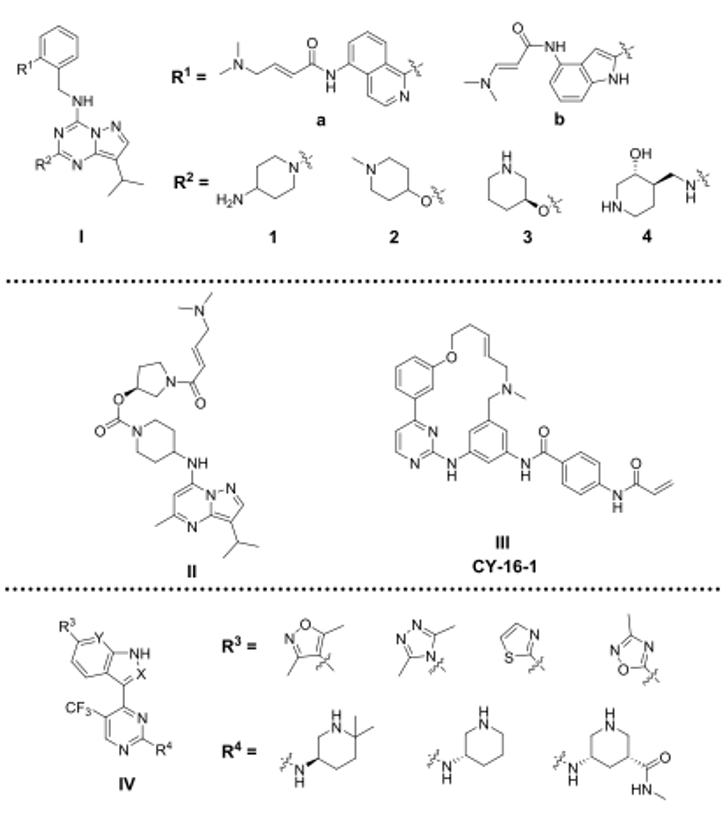
图7. 在研中的新型CDK7抑制剂化学结构
UD-017是一种取代的二氢吡咯并吡唑衍生物,在小鼠中具有50%的口服生物利用度和6.8 h的t1 / 2表现出可逆、选择性的CDK7抑制作用[85,106,107]。对CDK1-6/9(见表1)与313种激酶具有很高的选择性[84]。CRC(HCT-116)移植瘤小鼠以25、50和100 mg/kg剂量每日一次持续口服给药14天,可减少肿瘤体积,没有体重减轻或骨髓抑制迹象[85]。多发性骨髓瘤(NCI-H929)移植瘤小鼠在50mg/kg剂量每天口服一次连续14天时,它几乎完全抑制了癌症的生长[88]。口服给予一系列实体瘤的PDX模型,包括NSCL癌(LXFL1121)、胃癌(GXA3067)、肉瘤(SXFS117)和胸膜间皮瘤(PXF541)时,它也会使肿瘤生长消退或抑制[86]。C-MYC的下调与体内CDK7抑制和抗肿瘤效果具有相关性。
YPN005是一种口服有效的CDK7抑制剂,已显示出有望治疗MYC依赖性癌症,包括肝癌和TNBC[108]。此外,Aurigene先前还公告称已开发出两种共价、选择性和可口服的CDK7抑制剂,其化学结构类型为吡唑并[1,5-a] [1,3,5]-三嗪或吡唑并[1,5-a]嘧啶衍生物和吡唑衍生物,它们的CDK7项目仍在进行中[109-111]。其中之一, Au-12122(CDK7 IC50 = 2 nM),当以30mg/kg每日一次或每日两次剂量给药时,在MV4-11小鼠异种移植模型中肿瘤生长抑制率分别为77%和87%[109]。没有观察到明显的体重减轻。TGN-1062是可逆CDK7抑制剂,在小鼠中具有62%的口服生物利用度。为了优化其对其他CDK的选择性,开发出了TGN-1069,目前正在对其进行激酶谱(kinome profiling)分析、ADME-TOX分析和药效研究[112]。QS1189化学结构为吡唑并三嗪的CDK7抑制剂,在体外可抑制源自各种淋巴瘤的肿瘤生长[113]。尽管QS1189在激酶谱分析中有相当的选择性,但与其他CDK相比,它对CDK7缺乏特异性。本节中所有抑制剂的结构目前均未被公开。
另一方面,一项专利审查揭示了其他化学骨架可能作为新一代CDK7抑制剂基础。然而,由于尚未公开上述抑制剂的结构,因此不能排除这些骨架中类似于上述抑制剂的可能性。新骨架的设计方法包括开发BS-181的不可逆衍生物,THZ1的共价多环衍生物和SY-1365的可逆衍生物(见图7)。与Samajdar等人[111]相似,各种专利探索了在保留LDC化合物的吡唑并[1,5-a]-[1,3,5]三嗪母核或BS-181和CT7001的吡唑并[1,5-a]嘧啶骨架的同时移植各种亲电弹头的方法[114−116]。Nam等人[116],保留了LDC3140的N-苄基吡唑并[1,5-a] [1,3,5] triazin-4-胺母核(图7,结构I),并探索了苄基氨基部分C2位的几个迈克尔受体型取代基(R1)和吡唑并[1,5-a] [1,3,5]三嗪(R2)的C2位置的各种官能团。亲电弹头通常通过酰胺连接臂(linker)与芳基/杂芳基/杂环烷基连接,所述芳基/杂芳基/杂环烷基进一步与苄氨基部分连接。在R1处通常为耐受性良好的杂芳基包括异喹啉(例如,R1 = a)和吲哚(例如,R1 = b)。苯氧基或哌啶基氧基衍生物也导致对CDK7的IC50小于100 nM。但是,异喹啉衍生物似乎赋予对CDK7优于CDK1/2/5的选择性,特别是当与R2与4-氨基哌啶基组合使用时,例如IR1 = a和R2 =1。R2取代基的其它可选基团包括已经优化过的片段,例如作为LDC3140中的1-甲基哌啶-4基-氧基、LDC4297中的(S)-哌啶-3-基氧基和CT7001的(3S,4S)-4-氨基甲基哌啶-3-醇,所有这些都具有对CDK7的良好抑制率和对CDK1/2/5的选择性。激酶组的选择性尚未可知。
结构II(图7)遵循上述相同的基于衍生物的设计方法,并含有吡唑并[1,5-a]嘧啶母核,但用4-氨基哌啶片段代替了前一系列的苄氨基片段[114]。吡唑并[1,5-a]嘧啶环在C5处的取代限于甲基。结构II对CDK7的IC50为93nM,ATP浓度为表观Km时,对CDK4/9的抑制作用较弱,IC50分别为2.83与6.32μM[115],对CDK1/2/6/8/12/13/18/19的IC50大于8μM,这证明对CDK家族具有良好的选择性,但尚未公开对激酶组的选择性。细胞生物测定也反映了对CDK7的抑制作用,结构II抑制了CTD处Ser5的磷酸化(IC50 = 0.148μM),但在测试HCT-116细胞Ser2磷酸化(CDK9底物)的抑制作用时,其IC50大于20 mM。它在体外抑制各种癌细胞系的增殖,包括抑制结肠、乳腺、肺、卵巢和胃癌细胞的增殖,IC50范围从0.14至0.48μM,在口服20mg/kg剂量时在异种移植瘤模型中显示出对这些肿瘤显著抑制作用。
新型CDK7抑制剂的设计也基于THZ1及其类似物的结构。Chen和Lou将大环化合物TG02(一种CDK、JAK2和FLT3抑制剂)[117]与THZ/SY系列的亲电子弹头组合,以获得共价CDK7抑制剂。其中就有化合物CY-16-1,见即图7的结构III[118]。CY-16-1抑制CDK7的IC50小于15 nM,Kd值小于1 nM,在体外抑制一系列SCLC细胞系,IC50范围为0.018-0.032μM。另一方面,一些专利考虑了保留THZ1和SY-1365的4-(1H-吲哚-3-基)嘧啶-2-胺母核或进一步用吲唑、1H取代吲哚、1H-吡唑-[3,4-b]吡啶替换后开发可逆的类似物[119-122]。在结构IV的R3处引入各种五元杂芳基(图7),并在R4处引入取代的哌啶-3-胺,导致对CDK7的IC50不到30 nM,而对CDK2/9/12具有良好的选择性(IC50大于500 nM)[121]。但是,这些化合物对同一癌细胞类型具有不同的抗增殖作用,IC50范围从低nM到高于1μM。这可能是由于它们对整个激酶组的抑制活性不同而导致的。总的来说,全球范围内对开发新的CDK7抑制剂的兴趣不断浓厚,这将推动新结构的开发,并很快进入临床试验。
总结与展望
一方面,CDK7独特地参与转录调节和细胞周期进程,另一方面,CDK7在许多类型的癌症中异常过表达。因此已经着手将CDK7抑制剂往治疗药方向开发,并在许多临床前模型中显示出有希望的抗癌活性。它们在抑制过度活跃的基因表达和破坏细胞周期进程方面的作用似乎为癌症的治疗提供了相当大的机会,特别是那些目前尚无治疗方法的癌症,因此提出了一种潜在的革命性治疗方法。目前的CDK7抑制剂的作用机制似乎取决于癌细胞的分子遗传学。例如,THZ1在治疗后24小时会损害MYCN扩增的神经母细胞瘤细胞的细胞周期进程,但即使在暴露48小时后,对MYCN未扩增的细胞的细胞周期影响很微小。另外,不同抑制剂的生物学作用有些不一致。THZ1通常引起细胞周期停滞并诱导细胞凋亡,而使用更具选择性的YKL-5-124则未检测到明显的细胞凋亡,这可能是由于它们具有不同的生化特性所致。因此,尽管在实现所需的选择性和药理学方面取得了重大进展,但靶向CDK7的抗癌药开发仍处于起步阶段。迄今为止,CDK7抑制剂已显示出令人信服的安全性以及较低的副作用,并且已针对各种晚期实体瘤的治疗方案已经就绪,以评估其作为单药用药和联合用药的临床疗效。这些结果对于确定临床前疗效是否可以转化为临床效果以便CDK7抑制剂可以闻到成功的甜味至关重要。
行业新闻
- 2024年12月9日,Recursion公司(RXRX)报告了其潜在的同类最佳CDK7抑制剂REC-617单药治疗的临时1期临床数据,显示出令人鼓舞的患者反应和良好的耐受性。REC-617就是湃隆生物(Apeiron)与Exscientia(EXAI)联合开发的GTAEXS-617;根据2024年6月18日EXAI的公告,EXAI收购了GTAEXS-617的全部权益,在并入RXRX之后,开发代码更名为REC-617。
- 2024年5月29日,Qurient在ASCO 2024年会上公开Q901的一期临床研究结果。Hao Xie et al., A first-in-human trial of selective CDK7 inhibitor Q901, in patients with advanced solid tumors: Interim results of a phase I study (QRNT-009).. JCO 42, 3078-3078(2024). DOI:10.1200/JCO.2024.42.16_suppl.3078
- 2024年5月14日,Clinicaltrials的网站显示Exelixis的XL102因为商业原因终止临床研究,参见:NCT04726332。
- 2024年3月12日,Clinicaltrials的网站显示Qurient公司开始其高选择性的CDK7抑制剂Q901对晚期实体瘤的临床研究,参见:NCT05394103。
- 2023年8月3日,亿腾景昂今日宣布,其CDK7抑制剂项目EOC237在江苏省人民医院成功完成I期临床试验首位患者给药。
- 2023年7月23日, CT7001(samuraciclib)一期临床研究结果公布:Coombes, R.C., Howell, S., Lord, S.R. et al. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat Commun 14, 4444 (2023). https://doi.org/10.1038/s41467-023-40061-y
- 2023年5月17日,同源康医药宣布,其自主研发的新一代口服、高效、高选择性的小分子CDK7抑制剂TY-2699a获得国家药品监督管理局药品审评中心(CDE)同意开展临床试验的正式函件。这是国内首款同时获得中国CDE和美国FDA批准临床试验的CDK7抑制剂。目前全球没有获批上市的CDK7抑制剂,庞大的临床需求仍未得到满足。
- 2023年2月17日:同源康医药CDK7抑制剂TY-2699a获得国内首个FDA临床批准
- 2023年2月13日,亿腾景昂首个自主研发项目CDK7抑制剂递交临床试验申请
- 2022年12月8日,Exelixis公布了其小分子CDK7抑制剂XL102在晚期实体肿瘤患者中的1期临床试验初步数据。
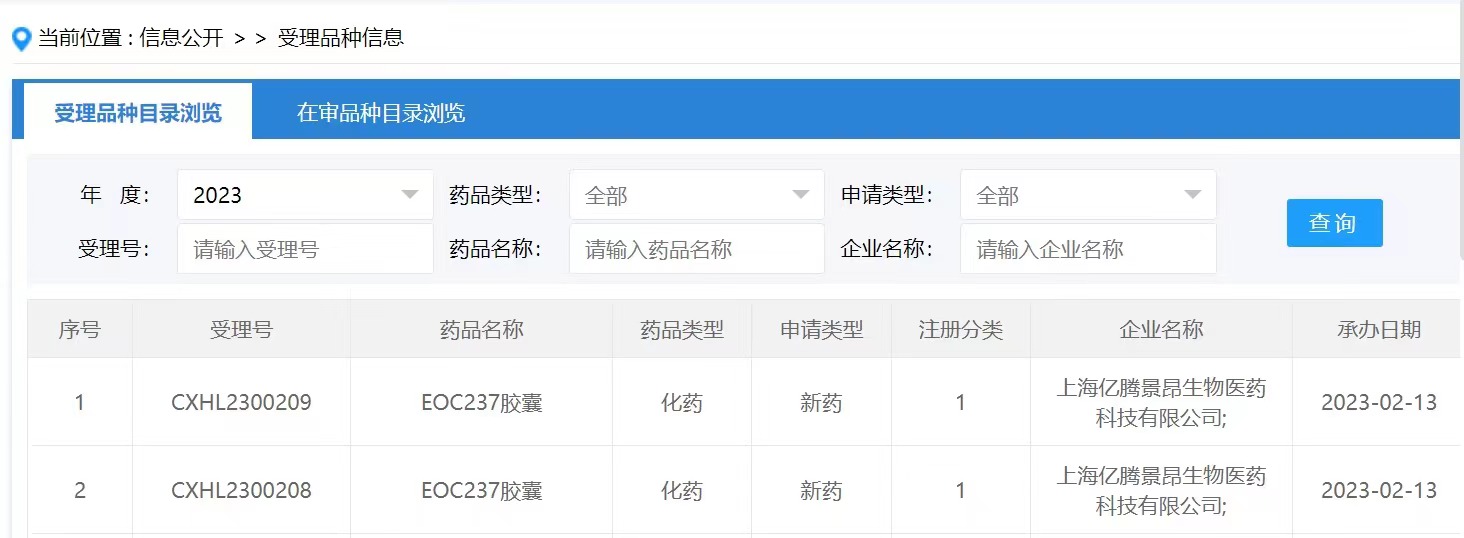
- 2022年6月30日,湃隆生物科技有限公司(香港)的CDK7抑制剂专利公开(WO2022134641A1),可以发现该专利要求的权利要求式I化合物以及实施例化合物与Syros公司2021年在J Med Chem上公开的SY-5102类似,具有非常一样的结构特征,如下图所示。
- 2022年6月29日,Theodora A. Constantin等人的研究结果表明,CT7001对阉割抗性前列腺癌(CRPC)有效。
- 2022年4月13日,Exscientia(EXAI)在2022年美国AACR年会上的poster(Abstract number: 3930)公开了其选择性CDK7抑制剂GTAEXS-617的数据。
- 2022年3月29日,广州费米子科技有限责任公司的CDK7抑制剂专利申请《嘧啶基衍生物、其制备方法及其用途》公开,中国专利申请号:2021111199080.0
- 2022年3月11日,Dana-Farber Cancer Institute Inc的CDK7蛋白降解剂中国专利申请《细胞周期蛋白依赖性激酶7(CDK7)的降解剂及其用途》公开,中国专利申请号:202080050815.4
- Syros公司在J Med Chem上发表文章,公开SY-5609的结构与数据。原文:Marineau, J. J.; Hamman, K. B.; Hu, S.; Alnemy, S.; Mihalich, J.; Kabro, A.; Whitmore, K. M.; Winter, D. K.; Roy, S.; Ciblat, S.; et al. Discovery of SY-5609: A Selective, Noncovalent Inhibitor of CDK7. J. Med. Chem. 2021. https://doi.org/10.1021/acs.jmedchem.1c01171.
- 2021年9月20日,Syros公司公开选择性CDK7抑制剂SY-5609的I期临床实验数据
- 2021年9月16日. Carrick therapeutics公司公布CDK7抑制剂CT7001(SAMURACICLIB)临床2a的耐受性与有效性数据。
- 2021年8月16日,Carrick therapeutics公司公布CDK7抑制剂CT7001(SAMURACICLIB)首批临床数据。
- 2021年8月2日,Carrick therapeutics公司公布与罗氏合作CDK7抑制剂CT7001(SAMURACICLIB)联合用药治疗HR+的评价。
- 2021年7月21日,专注于创新肿瘤药物研发的上海湃隆生物科技有限公司与英国的制药科技公司Exscientia共同宣布:旨在针对性治疗由异常细胞周期引发的癌症,建立一个包含多个特异性靶向不同CDK的创新产品管线。…双方合作深化是建立在此前成功合作设计高选择性CDK7抑制剂的基础上开展的。
- 2021年4月16日,浙江同源康医药股份有限公司管线上的CDK7抑制剂TY-2699相关中国发明专利申请CN202110129797.5公开。该专利以Syros公司的专利WO2020093006实施例101化合物为参比,估计同源康TY-2699是对SY-5609的跟进。据同源康的管线介绍,TY-2699目前处于IND阶段中,估计即将进入临床阶段。
- 2021年1月27日,Exelixis的CDK7抑制剂XL 102临床试验在ClinicalTrails登记:NCT04726332
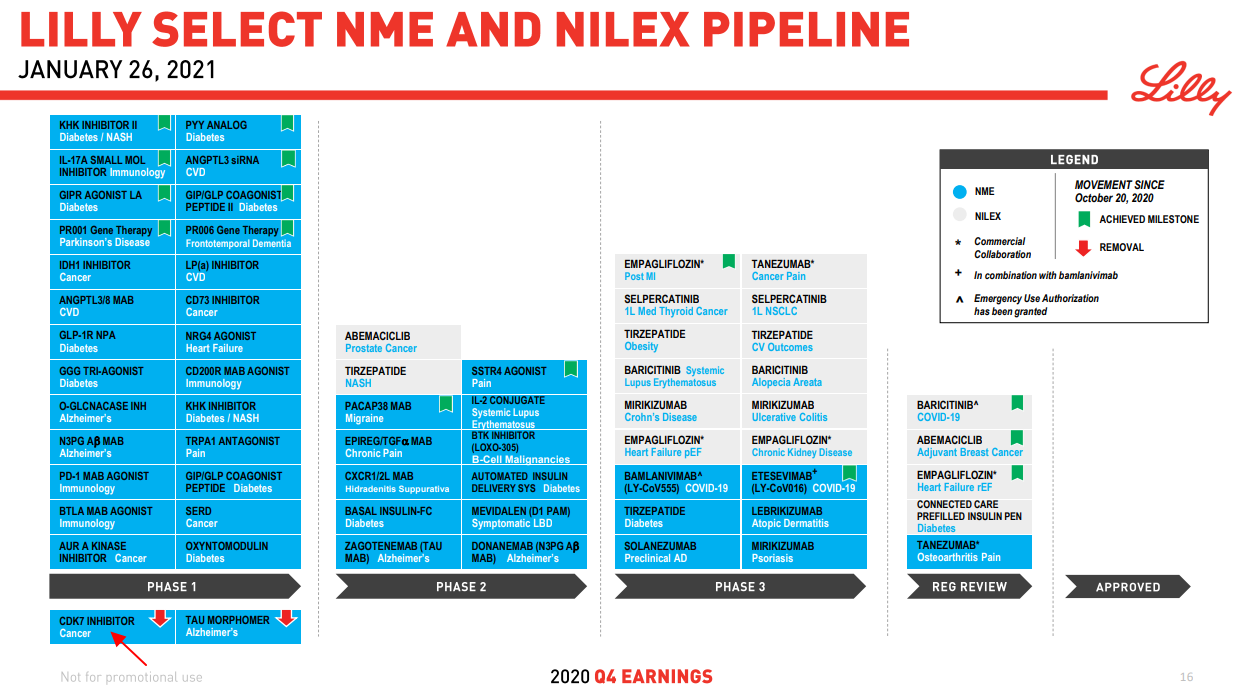
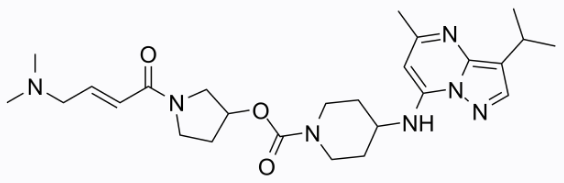
- 2021年01月26日,Lilly将CDK7抑制剂LY-3405105从其研发管线中移除,官网肿瘤管线也看不到该项目的信息。
- 2021年01月25日. CT7001与CDK7的复合物晶体结构被解释出来。Greber BJ, Remis J, Ali S, Nogales E. 2.5Å-resolution structure of the human CDK-activating kinase bound to the clinical inhibitor ICEC0942. Biophys J. 2021 Jan 18:S0006-3495(21)00047-3. doi: 10.1016/j.bpj.2020.12.030. Epub ahead of print. PMID: 33476598.
- 2021年01月25日. 据报道,Exelixis公司开始其CDK7抑制剂XL-102的临床实验,适应症为晚期实体瘤或者转移性实体瘤,临床将扩展到卵巢癌、乳腺癌与前列腺癌。
- 2020年12月9日. Exelixis引进Aurigenes开发的共价、选择性CDK7抑制剂AUR102/XL102
- 2020年10月24日. Syros披露SY-5609的I期临床实验初步数据
- 2018年12月10日. 礼莱CDK7抑制剂LY-3405105开始Phase 1a/b临床实验
- 2017年11月30日. Carrick therapeutics公司宣布CDK7抑制剂CT7001入组第一位I期临床受试者
2023年2月15 日,同源康医药宣布,其自主研发的新一代口服、高效、高选择性的小分子CDK7抑制剂TY-2699a获得美国食品药品监督管理局(FDA)同意开展临床试验的正式函件(Study May Proceed Letter)。这是同源康医药第三个获得FDA批准临床试验的创新药。
TY-2699a是国内首款获得美国FDA批准临床试验的CDK7抑制剂。目前全球没有获批上市的CDK7抑制剂,仅有4款药物处于早期临床试验阶段(均为I/II期),国内尚无进入临床开发阶段的研究药物,庞大的临床需求仍未得到满足。
同时,同源康医药已与国家食品药品监督管理局(NMPA)药品审评中心(CDE)进行了TY-2699a的Pre-IND沟通交流,将于近期正式提交IND,有望于2023年5月获得NMPA批准的临床试验通知书。
2月13日,亿腾景昂药业,一家在肿瘤创新药领域集药物发现、研究、开发和商业化为一体的生物科技公司宣布,已经向国家药品监督管理局药品审评中心(CDE)正式递交了EOC237的临床试验申请并正式获得受理。
2023年,EOC237将进入I期临床试验,拟定适应症为晚期实体瘤,因目前国内尚未有同靶标分子递交临床试验申请,EOC237有望成为国内首个进入临床试验阶段的CDK7抑制剂。
截至2022年9月7日的数据,26例患者接受了治疗。 1例乳腺癌患者和1例脂肪肉瘤患者达到SD。XL102在评估的剂量水平下耐受性良好。
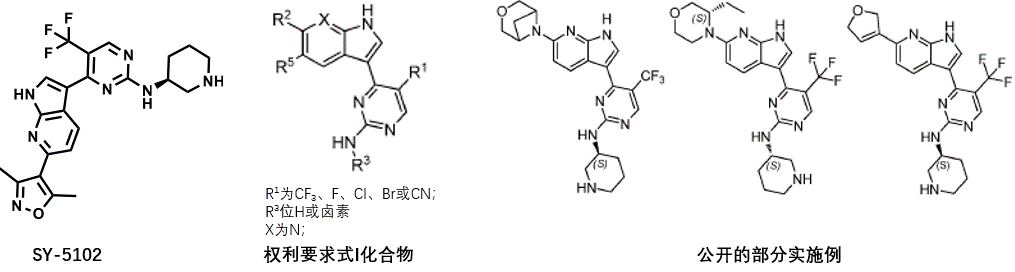
WO2022124641A1专利公开的方案以SY-5102为起点,主要对异恶唑环或哌啶环进行进行替换或修饰。
此前湃隆生物与Exscientia(EXAI)在2022年美国AACR年会上公开其由AI设计的选择性CDK7抑制剂GTAEXS-617的临床前数据,鉴于湃隆生物近期并没有其它相关的专利公开,让人疑心WO2022124641A1即是GTAEXS-617相关的专利。果真如此的话,该项AI设计也只是药化常规的me-too设计。
研究表明,CT7001选择性地与前列腺癌细胞中的CDK7接触,引起增殖抑制和细胞周期停止。p53的激活、细胞凋亡的诱导、全长和组成型活性AR剪接变体介导的转录抑制有助于体外的抗肿瘤疗效。口服CT7001可抑制阉割抗性前列腺癌(CRPC)异种移植瘤的生长,并明显增强恩扎鲁胺的生长抑制作用。对接受治疗的异种移植体进行的转录组分析表明,细胞周期和AR抑制是CT7001在体内的作用模式。目前抑制雄性激素受体(AR)的策略对CRPC没有治疗效果。细胞周期蛋白依赖性激酶7(CDK7)除了在细胞周期和全球转录调控中发挥既定作用外,还促进了AR信号的传递,这为CRPC的治疗目标提供了依据。
Constantin, T. A.; Varela-Carver, A.; Greenland, K. K.; de Almeida, G. S.; Penfold, L.; Ang, S.; Ormrod, A.; Ainscow, E. K.; Bahl, A. K.; Carling, D.; et al. The CDK7 Inhibitor CT7001 (Samuraciclib) Targets Proliferation Pathways to Inhibit Advanced Prostate Cancer. bioRxiv 2022, 2022.06.29.497030. https://doi.org/10.1101/2022.06.29.497030.
湃隆生物官网在2022-3-18对此报道:湃隆生物高级副总裁Fred Aswad博士将出席于2022年4月8日至13日在美国新奥尔良举行的AACR会议,并做专题演讲。这次演讲的内容将是关于GTAEXS-617的临床前数据。GTAEXS-617是一种借助人工智能研发的口服、高效和高选择性的CDK7小分子抑制剂,目前正在进行IND研究。临床前数据显示,GTAEXS-617具有良好的成药性,在荷晚期高级别浆液性卵巢癌和三阴乳腺癌的异种移植瘤小鼠中具有强大的抗肿瘤活性,导致肿瘤完全消退。通过利用Exscientia的精确肿瘤学平台,湃隆生物与Exscientia发现GTAEXS-617对原发性卵巢癌患者样本的不同影响,希望借此定义一个患者选择生物标记物,以筛选更有可能对CDK7抑制产生响应的患者。
从公开的实施例可以看出,该专利的技术方案以Syros的SY-5609为基础进行了模仿,部分实施例如下:
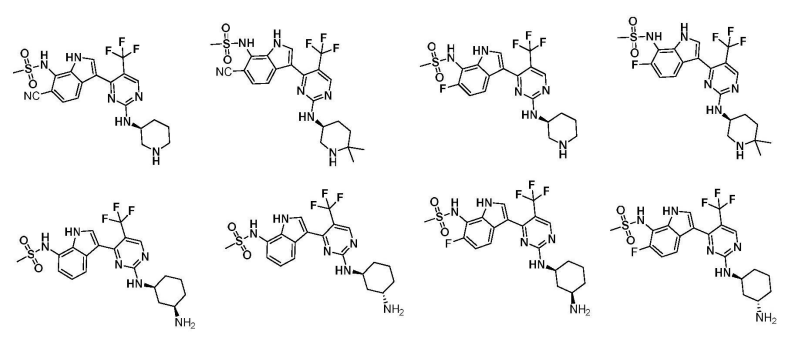
国际公布号为WO2022063212A1的同族世界专利申请检索报告认为:1)权利要求1-12,28-45没有新颖性与创造性;2)权利要求13-27没有创造性。比较有意思的是,检索报告的主要依据是SYROS制药公司在先的专利公开WO2020093006A1。很明显,广州费米子的CDK7抑制剂权利要求授权前景存在很大程度的不确定性。

根据广州费米子官网介绍,该公司的核心技术是AI分子设计:运用AI算法根据蛋白口袋形状和性质要求,基于骨架跃迁、侧链替换、电子等排等策略,让计算机自动生成目标化合物库,库中的新分子具有良好的类药性和可合成性。从官网上找不到CDK7抑制剂相关的开发进展信息,也找不到任何药物开发的管线与相关信息。
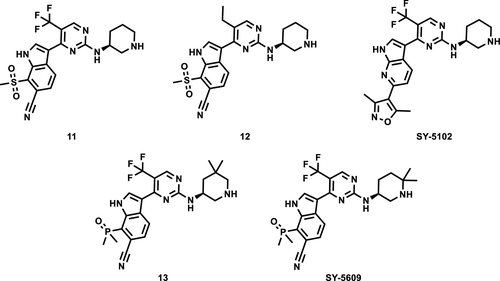
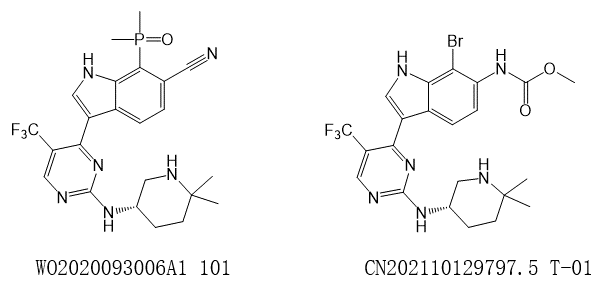
图1S. WO2020093006的代表性化合物以及101CN202110129797.5的代表性化合物T-01

根据CT7001与CDK7以及CDK2的共晶结构(PDB code分别为7B5Q与5JQ5),对CT7001的构象稳定性进行分析,发现CT7001与CDK2的结合构象自由能要高于与CDK7的结合构象自由能,这可能是CT7001具有选择性的原因之一。具体见:CT7001的CDK7/CDK2激酶靶标选择性分析。
LY3405105的化学结构式


