
摘要:本文分享了Mould等人如何从高通量筛选发现苗头化合物、再用骨架跃迁进行苗头化合物的优化、结合文献信息进一步修饰化合物与母核、最终发现活性保留、ADME与毒性改善的先导化合物。
Mould, D. P.; Alli, C.; Bremberg, U.; Cartic, S.; Jordan, A. M.; Geitmann, M.; Maiques-Diaz, A.; McGonagle, A. E.; Somervaille, T. C. P.; Spencer, G. J.; Turlais, F.; Ogilvie, D. Development of (4-Cyanophenyl)glycine Derivatives as Reversible Inhibitors of Lysine Specific Demethylase 1. Journal of Medicinal Chemistry 2017, 60:7984-7999. DOI:10.1021/acs.jmedchem.7b00462

一. 背景
抑制赖氨酸特异性组蛋白去甲基化酶1(Lysine specific demethylase 1, LSD1)可以诱导急性髓细胞样白血病(acute myeloid leukemia,AML)的白血病干细胞分化。因为LSD1底物结合口袋不仅尺寸大、而且极性强,所以LSD1抑制剂的开发并非易事。
LSD1可逆抑制剂GSK-690在酶学测试与细胞测试中显示出良好的生物学活性,然而这个化合物同时也强烈的抑制hERG心肌离子通道,这阻碍了该化合物进一步进入临床的可能。还有许多文献也报道了可逆抑制剂,但是多有显著的脱靶作用以及非特异性效果。最近几年,发现该靶点引起了商业公司的兴趣,出现不少的专利申请,见图1。

图1. GSK-690(1)以及Quanticel制药公司的几个专利文献化合物(2-6)
二. 发现过程
(一) 高通量筛选发现苗头化合物
采用TR-FRET(time-resolved fluorescence resonance energy transfer)测试方法对LSD1高通量筛选了含有150000化合物的数据库。如果一个化合物对LSD1的抑制率(测试浓度为30μM,)大于70%,则认为该化合物是苗头化合物。其中0.3%的化合物为苗头化合物(472个),对这些化合物进一步进行了TR-FRET干扰性测试,并进行7点、12点的IC50测试,结果发现72个化合物的IC50值在4-120μM之间。
(二)苗头化合物的优化
对部分化合物重新合成、验证,选择看起来很有潜力的磺酰胺类化合物为起始化合物合成了一系列化合物,最后将化合物10a的活性从μM水平优化到10b的亚μM水平(见图2)。

图2.化合物10b的开发与特性。(A) 生化测试; (B) 细胞水平测试结果; (C) SPR测试结果.
不幸的是,10b虽然在生化测试中IC50达到0.43μM,但是此类化合物在细胞水平测试中并没有表现出活性,并且在SPR实验中也没有显示与LSD1的结合性能。此外,在与Tranylcypromine的竞争性结合实验中磺酰胺类化合物也没有显示出与LSD1相互作用的性能。其它的几个HTS命中的化合物在SPR测试中也没有表现出活性。这表明生化测试可能存在假阳性。
幸运的是高通量筛选确实也发现了一个化合物(12)可以可逆地与LDS1结合(图3),这个化合物可以看成是化合物1(图1)吡啶环的开环生物等排体替换。而化合物1由GSK公司在AACR2013年会由GSK首次披露,直到2015年仅有非常少的信息公开。为了验证磺酰胺类化合物是潜在的LSD1抑制剂新骨架,作者用Cresset SPARK与Torch对化合物进行了分子叠合、搜索片段库用具有相似空间与静电的骨架进行骨架跃迁。
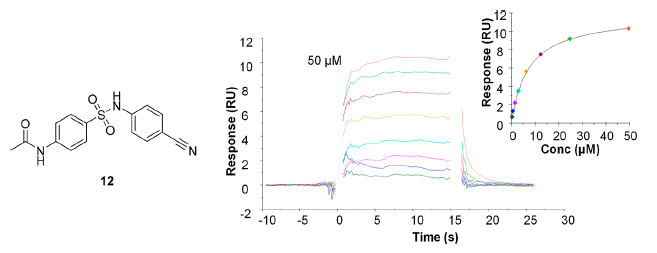
在输出的结果中,作者设计了初始的目标化合物一系列磺酰胺化合物(图5),具有更容易的合成可行性而与原型化合物的2D相似性更低。
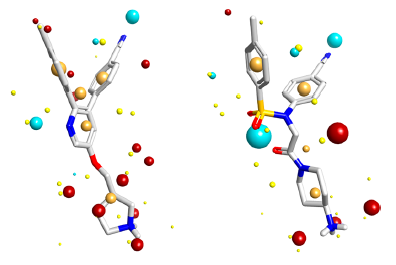
图5.化合物1(左)与16g(右)用Cresset Torch的叠合。相似性分值为0.712。
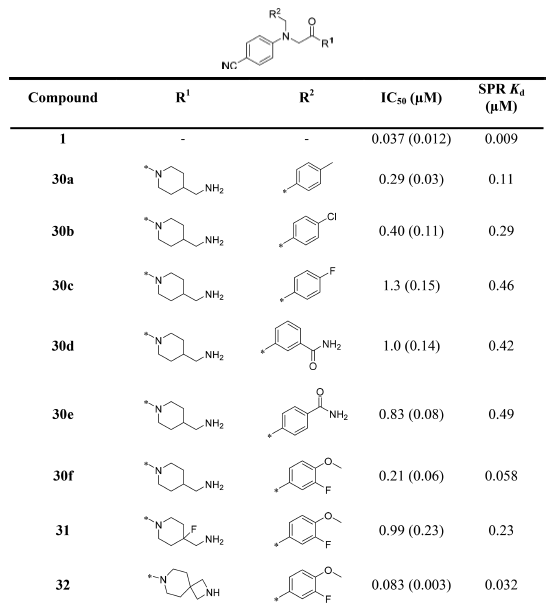
作者保留了化合物1的对甲苯磺酰基与对氰基苯基,聚焦于优化母核部分得到化合物16a-m(表1)。
表1.优化的新骨架

此类化合物虽然提高了活性,但似乎到也达了一个平台:没有一个化合物的Kd小于2μM。恰巧这时候,Quanticel公司的专利出现了,并带来了新的信息:5、6元环或5、6元双环母核化后展示出小于100nM的IC50活性(见图1化合物2-6)。对这些专利化合物进行分析,发现了必要化合物特征与可改造位置的提示。比如,在专利化合物里对位-氰基苯基与碱性N原子是保守的,而对甲基苯基-片段是可改造的。在此认识的指导下,作者又进行了结构修饰与活性测试,结果见表2。
表2. 对芳环进行修饰
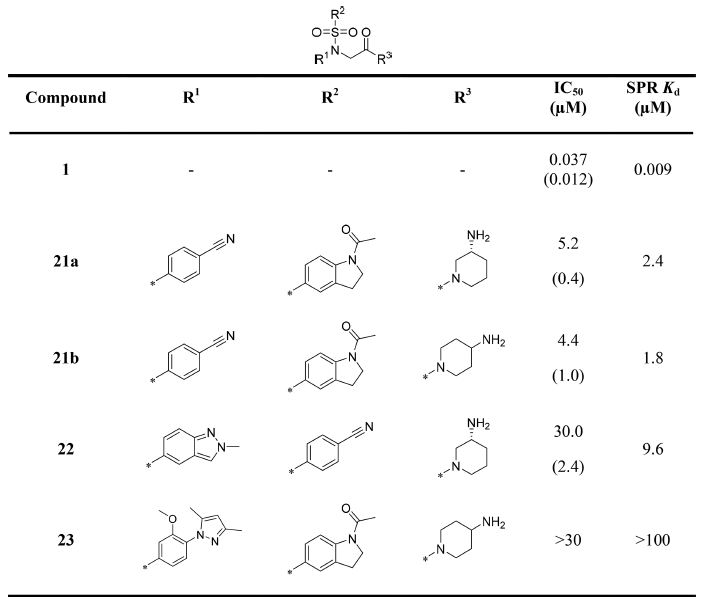
然而结果让人失望,没有一个化合物的活性获得显著地提高。因此作者打起了修饰母核的主意:改变母核是否可以提高活性?于是作者尝试将-SO2-替换为-CH2-,结果见表3。
表3. 对(4-Cyanophenyl)glycine芳环的优化
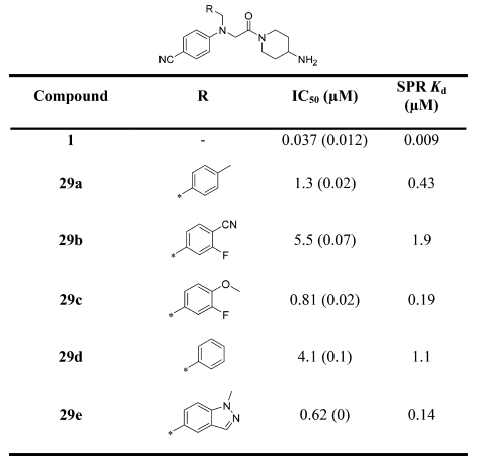
这个母核变化不仅显著地降低了分子量、而且减小了分子极性表面积,这有利于化合物的膜渗透性与生物利用度。然而,也因为引进了苄基可能使得化合物具有代谢稳定性的问题。首先,作者采用了化合物16g的4-氨基哌啶作为碱性中心着眼于修饰甲苯基。比之16h,化合物29a在IC50与SPR实验上获得了5倍的活性提升。此外,苯环也耐修饰,比如29c化合物也获得了几倍的活性提升。
作者又研究了碱性中心对活性的影响,发现将4-氨基哌啶-单元改为4-胺甲基哌啶-提高4倍的活性(比如29a vs 30a,见表4)。其中引入螺环进行柔性约束的化合物(32)活性提升至IC50小于100nM。
表4. 对碱性氨基的优化
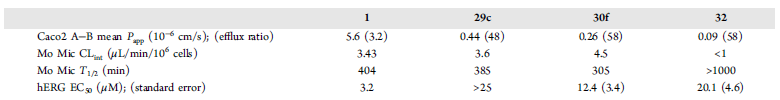
化合物29c、30f与32对hERG离子通道IC50得到改善(表5),这些化合物提高了靶点选择性。
表5. 化合物1、29c、30f与32的ADME/Tox性质
三. 小结
- 作者的第一步是通过高通量筛选获得苗头化合物
- 选择有潜力的苗头化合物用SPARK进行骨架跃迁
- 骨架跃迁计算需要结合文献信息进行调整,信息用的越多,越有利于朝目标靠近。
- 对母核进行了一个小小的修饰,最后发现对LSD1的抑制活性得到提高而对hERG抑制得到降低的先导化合物。
SPAKR的骨架跃迁技术在本研究中提供了新母核。虽然SPARK直接推荐的化合物活性并不理想,经过几次的试错,作者最后发现了比较理想的先导化合物。这提醒我们,骨架跃迁计算的首要目的是获得新骨架的灵感,而不是依赖软件生成最终理想的化合物。
借助专利文献,作者引入新的假设,进一步遍历了化合物空间,发现活性改进的余地不大,认为需要进一步修饰母核。


