
选择性METTL3抑制剂EP652苗头化合物1的发现
摘要:METTL3抑制剂苗头化合物 1 的发现过程综合运用了基于配体的药物设计(LBDD)、结构生物学(SBDD)、QSAR 建模和骨架跃迁等策略。从初始的靶点挑战到化学起点的识别,再到假设验证和新骨架的开发,最终通过实验确认了其高效性和特异性。这一过程展示了Cresset Discovery利用计算化学工具Spark、Flare、3D-QSAR、EC等工具的强大能力,为后续优化(如 EP652 的发现)奠定了基础。
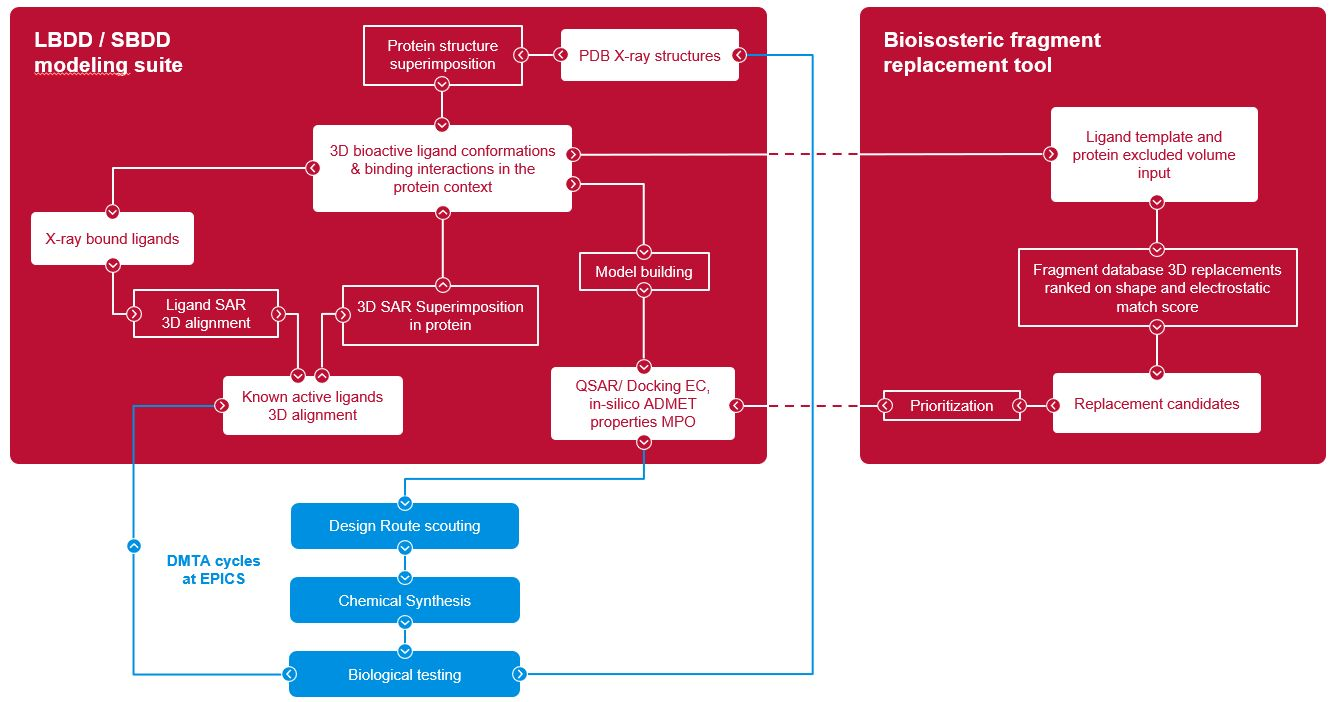
背景与挑战
METTL3是一种RNA甲基转移酶,主要负责N6-甲基腺嘌呤(N6-methyladenosine,m6A)的加成反应,这是mRNA中最丰富的修饰。m6A的普遍存在以及METTL3的活性和表达与急性髓系白血病(AML)的出现和进展相关联,使得METTL3成为癌症治疗极具吸引力的靶标1,2。
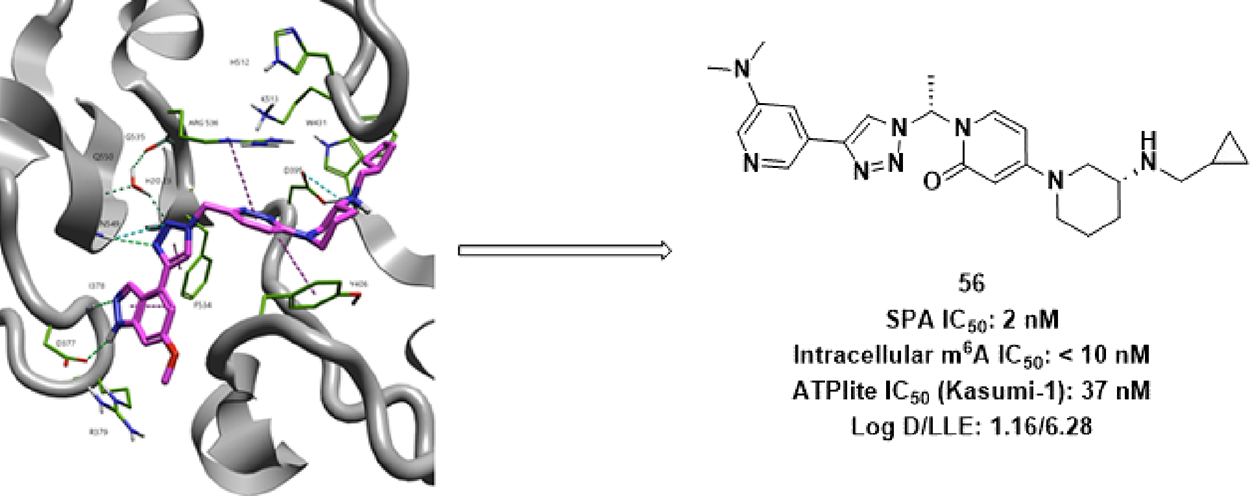
图1. METTL3抑制剂EP652(化合物56)
最近,Epics Therapeutics AS公司的Dutheuil等人3报道了小分子METTL3抑制剂的发现和优化过程,最终选定EP652(图1,化合物56)作为体内概念验证化合物。EP652能够强效抑制METTL3的酶活性,具有良好的药代动力学(PK)参数,并在临床前肿瘤学模型中表现出疗效,表明药理学抑制METTL3是治疗液态和实体瘤的可行策略。
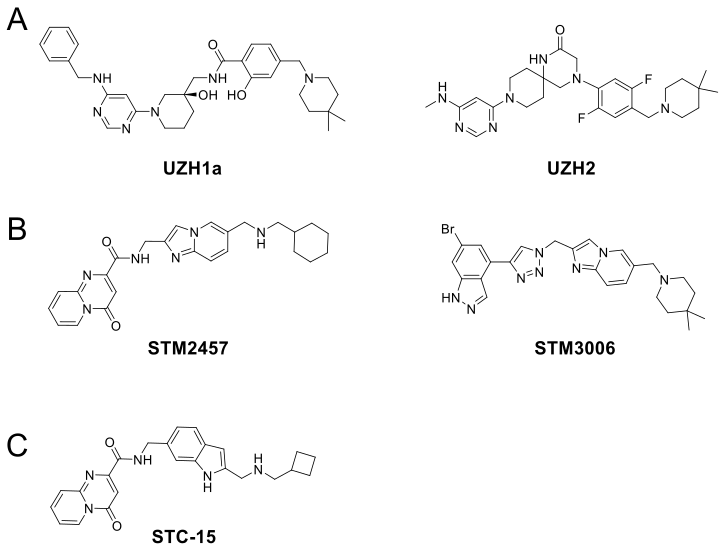
图2. 文献报道的METTL3/14抑制剂
然而,在项目开始时,尚无选择性的 METTL3 抑制剂,图2给出了一些文献报道的非选择性的METTL3抑制剂。已知的参比化合物(如 sinefungin)是非选择性的,会抑制大多数甲基转移酶。因此,研究团队面临的主要挑战是找到一种特异性抑制 METTL3 的小分子化合物。其中苗头合物1(见图4)的发现对于EP652的发现与优化至关重要,因此本文的主要目的是介绍苗头化合物1的发现过程。
并行策略的探索
在项目初期,同时开展了高通量筛选(HTS)、虚拟筛选和基于配体的药物设计(LBDD),以寻找化学起点。
- 高通量筛选(HTS): 使用 MTase-Glo 和亲和选择-质谱(ASMS)测定进行筛选。然而,由于初筛命中率低、非特异性配体亲和力高以及检测干扰发生率高等问题,这一方法未能产生可进行苗头化合物扩展(hit expansion)的化合物。
- 虚拟筛选: 与 HTS 类似,虚拟筛选4也因类似问题未取得成功。
- 基于配体的药物设计(LBDD): LBDD 成为关键方法,得到了晶体学证据的支持。
简而言之,HTS和虚拟筛选方法4未能获得能够通过苗头化合物扩展(hit expansion)的化合物,原因是初步筛选命中率低,非特异性配体亲和力及检测干扰发生率较高。与此同时,已有文献报道的小分子抑制剂(如 UZH1a 和 STM2457,见图2)与截短的 METTL3/14 结合的复合物晶体结构(PDB 代码:7ACD 和 7O2I)为LBDD研究提供了基础,从而能够利用基于结构的设计(SBDD)来补充基于配体的设计方法。
在METTL3抑制剂的LBDD设计中,Cresset Discovery5不只是提供专业的药物设计CRO服务,同时还是计算建模的真正合作伙伴。
关键假设与建模
研究团队提出了一个核心假设:有效的 METTL3 抑制剂应同时利用酶的两个关键结合位点,即 SAM(S-腺苷甲硫氨酸)结合域的腺苷部分和 RNA 底物识别位点。Cresset Discovery5对这些体系的建模(图3-上)表明,小分子抑制剂与这两个位点的相互作用与观察到的构效关系(SAR)一致。这一假设得到后续发布的X-衍射晶体学数据(PDB 7O2I)的支持6-8。
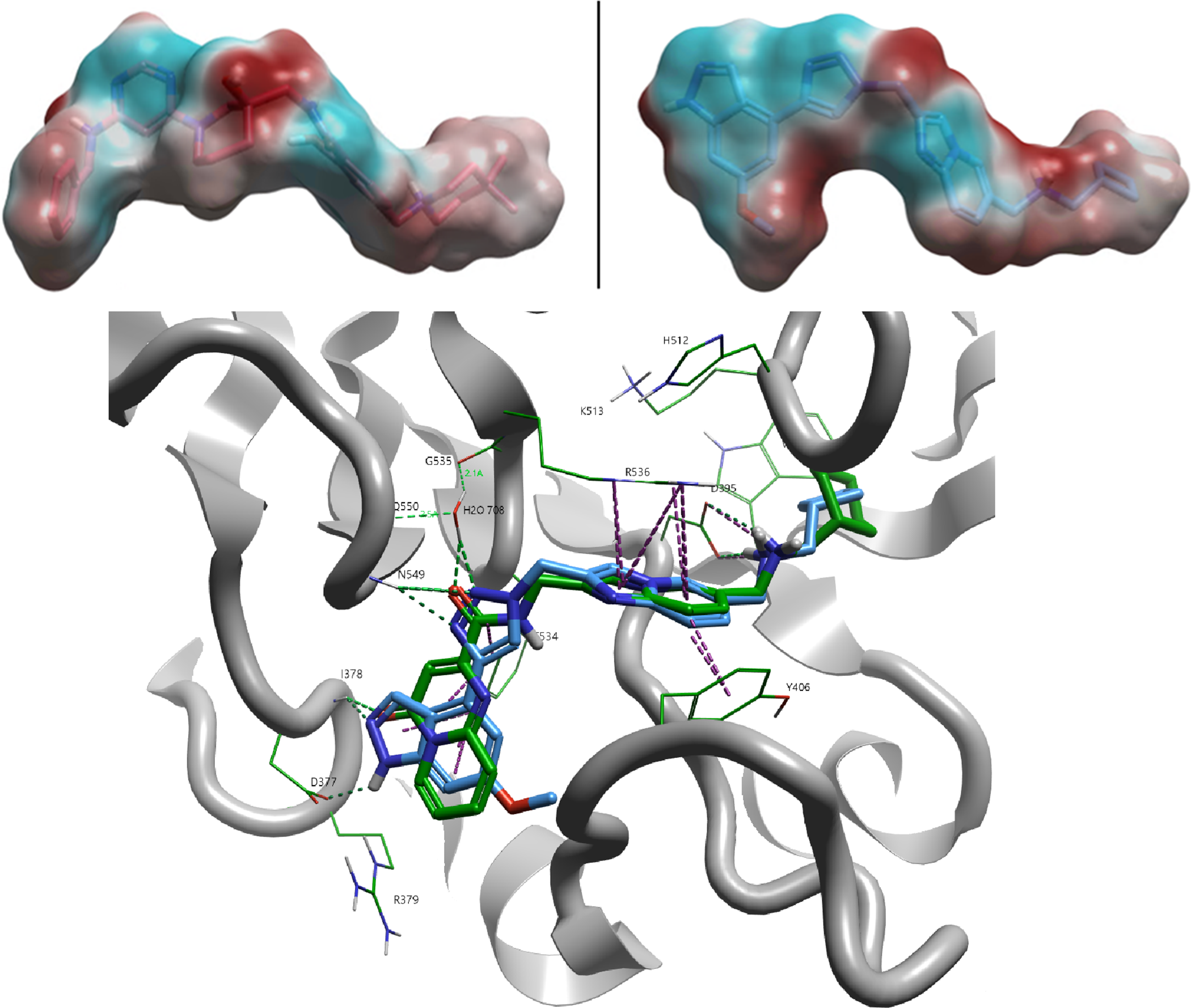
图3. 上图:UZH1a(左)和Storm#270(右)的静电表面比较,负静电场为蓝色,正静电场为红色。下图:Storm#270(蓝色)与STM2457(来自PDB:7O2I,绿色结构)的子结构叠合。氢键、阳离子-π和π-π堆积相互作用以虚线高亮显示。
对配体-酶相互作用的理解完善了我们的方法,通过分子对接寻找替代骨架。对参比化合物UZH1a和Storm Therapeutics化合物270(专利编号,STM3006的OMe变体)的场分析显示,如图3-上所示,两者在电荷分布和空间重叠上有显著相似性,并且在特定区域存在清晰的正负场分布。
对Storm Therapeutics专利中的关键子系列,包括三氮唑-吲唑类(如STM3006)和酰胺吡啶并嘧啶酮类(如STM2457),进行扩展分析,并将其对接至我们的模型(图3-下),团队得出结论,这些结构的母核单元主要是芳香连接臂(具有额外的π-π堆积和阳离子-π相互作用),连接两侧的可旋转基团(rotulas),从而允许在一侧的芳香头部基团(图4)以及在另一侧的氨基烷基基团与酶发生正确的相互作用。Cresset Discovery进一步提出,三氮唑-吲唑和酰胺吡啶并嘧啶酮片段应被视为一个整体解释为统一的“铰链”片段模式,而不是各自独立的功能单元(图4)。
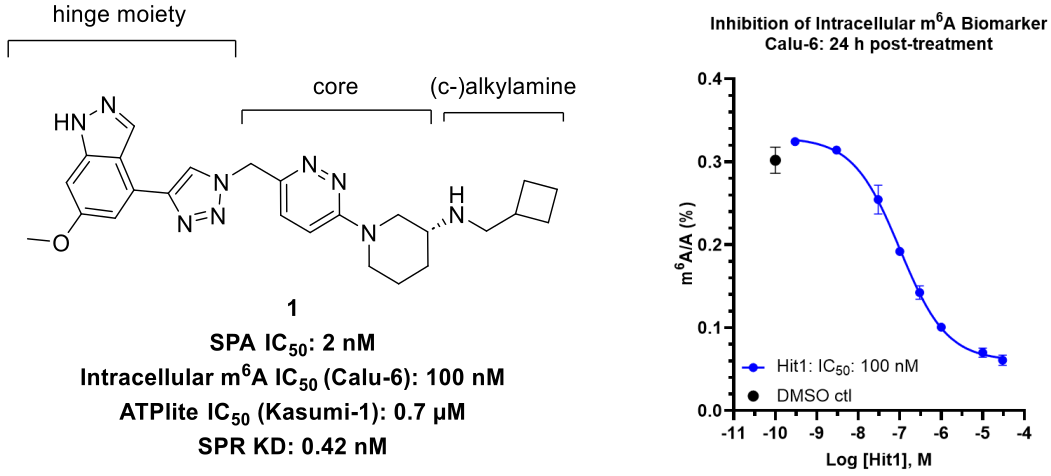
图4. 起始的苗头化合物1是通过计算机辅助母核替换获得的,并展示了代表性的抑制数据,突出了在用1处理Calu-6细胞24小时后,通过LC-MS/MS对mRNA中m6A的细胞定量结果。
基于这些假设和发现,我们开发了一个定量构效关系(QSAR)模型,从而通过计算机模拟进行骨架跃迁,旨在识别具有预测的改善ADME/PK特征9,10的新型骨架。我们的建模方法和所用工作流程的见下一小节。本次骨架跃迁使用了Cresset专有的片段替换工具Spark11,12。对Spark设计的结果使用多个精选的选择标准进行评估,包括分子对接13和静电互补性(EC)14,15,预测靶标亲和力,同时还使用理化性质标准(例如MW、LogP、TPSA和柔性)以及合成可行性评估来对8个概念分子进行合成优先级排序,其中之一是化学苗头化合物1(图4,活性最强的设计)。
模拟方法与计算流程
计算机模拟工作首先由Cresset Discovery对已发表的配体和METTL3的晶体数据进行详细分析,使用了Cresset公司的专有工具。利用Flare生成了3D蛋白质叠合和配体3D叠合,从而在与METTL3/METTL14体系所有可用蛋白质结构相同的坐标系中创建了一个完全叠合的构效关系(SAR)集合。这使得能够深入了解配体-蛋白质接触范围、配体形状、配体静电以及与活性化学结构相关的结合口袋中的水模式,以及不同的蛋白质loop结构。对于具有足够例子的化学结构,还可以从配体叠合和活性数据构建预测性的QSAR模型。这为对文献化学结构进行骨架跃迁设计进行优先级排序奠定了坚实的基础。我们使用Spark中的片段替换算法,该算法捕获参比生物活性构象的3D形状和静电特征,发现了一些潜在的新型生物等排体,但不具备2D结构相似性的可替代骨架候选化合物。对这候选化合物在蛋白质的背景下构建了3D结构,并使用诸如QSAR、3D相似度评分或生物等排体因子、对接评分和口袋静电互补性(EC)打分之类的上述指标进行表征。对那些具有合适的理化性质(即计算机计算的sLogP、TPSA)并且易于化学处理的候选化合物进行合成并测试活性。最终,这导致了选择活性苗头化合物系列,并对其进行进一步的类似物修饰。
一些最有前景的类似物通过X-衍射晶体学证实了其结合假设,令人惊讶的是,在环丙基取代的胺类化合物中观察到新的环状结构。相反,环丁基类似物遵循其他文献示例化学结构中观察到的现有模式。通过仔细检查晶体结构可以明显看出,与Trp431发生相互作用的是Storm Therapeutics和Caflisch化学结构SAR相关的关键组成部分,并且与靠近loop的位移是一致的。然而,环丙基类似物足够小,可以容纳另一种loop构象,同时与Trp431发生相似的相互作用,但具有几何上更紧凑的loop结构。这个高度柔性的loop及其悬垂残基的整体轨迹受到这个微小变化的影响。目前尚不清楚这是否具有任何实际的药理学意义,优势或劣势。
图5. METTL3抑制剂苗头化合物发现计算工作流
实验验证
这个优选的苗头化合物1在SPA初筛、多种细胞系的mRNA m6A的浓度依赖性抑制、在各种细胞系(MOLM-13、Calu-6、Caov-3、KG1a、A549、SK-OV-3、FaDu、Kasumi-1、MV-4−11)处理72小时后,在细胞活力检测(ATPlite)表现出有效抑制癌细胞增殖的效果。
表1. 化合物1的实验验证
| 测定方法 | 结果描述 |
|---|---|
| 初筛 | 在SPA初筛中表现出极高的活性,IC50 = 2 nM。 |
| 细胞实验 | 表现出mRNA M6A浓度依赖性抑制。通过测量在多个细胞系(如 Calu-6、A549、SK-OV-3、FaDu、Kasumi-1)中 mRNA M6A的浓度,进一步确认对METTL3/14的抑制作用。例如,在Calu-6细胞中,IC50 = 100 nM。 |
| 抗增殖作用(ATPlite) | 在细胞活力测定中,化合物 1 在多种细胞系(如 MOLM-13、Calu-6、MV-4-11 等)经 72 小时处理后显示出显著的抗增殖效果。例如,在 Kasumi-1 细胞中 IC₅₀ 为 0.7 μM。 |
| 晶体学验证 | X-衍射晶体学(分辨率 2.1Å)确认了化合物1 在 SAM 结合位点的结合模式,与预期一致。其共晶结构与已发表数据(Caflisch 课题组的 7O2F 和 Storm Therapeutics 的 7O2I)高度重叠,进一步验证了设计假设。 |
进一步通过X-衍射晶体学(2.1 Å分辨率)验证并表征了先导化合物与METTL3/14复合物的结合,证实了化合物1与SAM结合位点的结合(图6)与预期一致16。
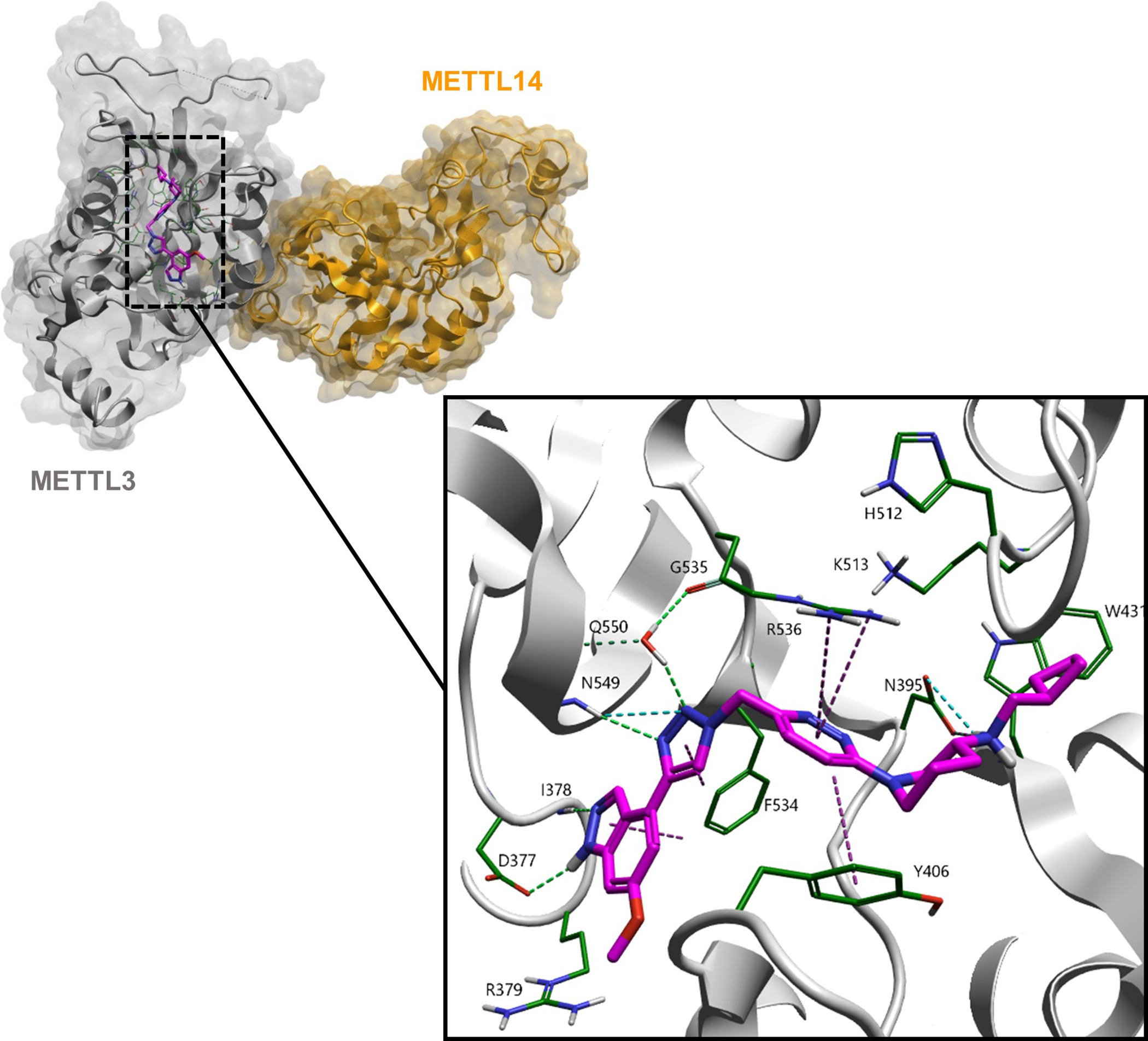
图6. METTL3:METTL14与1的复合物结构
获得1的共晶结构表明,活性结合位点与Caflish课题组最近发表的数据(PDB 7O2F与UZH2)和Storm Therapeutics的数据(PDB 7O2I与STM2457)非常吻合,这进一步验证设计假设。
结论
苗头化合物 1 的发现过程综合运用了基于配体的药物设计(LBDD)、结构生物学(SBDD)、QSAR 建模和骨架跃迁等策略。从初始的靶点挑战到化学起点的识别,再到假设验证和新骨架的开发,最终通过实验确认了其高效性和特异性。这一过程展示了Cresset Discovery利用计算化学工具Spark、Flare、3D-QSAR、EC等工具的强大能力,为后续优化(如 EP652 的发现)奠定了基础。
联系我们
想在自己的项目中亲自使用SPARK进行生物等排体替换,请联系我们获取免费的试用版;或者联系我进行在线演示;你还可以采购软件或委托研究与我们进行项目合作。
- 电邮:info@molcalx.com
- 电话:020-38261356
文献
- Vu, L.P. et al. (2017) “The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells,” Nature Medicine, 23(11), pp. 1369–1376. Available at: https://doi.org/10.1038/nm.4416.
- Zhang, Y. et al. (2023) “[Corrigendum] METTL3‑mediated m6A modification of Bcl‑2 mRNA promotes non‑small cell lung cancer progression,” Oncology Reports, 49(4), p. 65. Available at: https://doi.org/10.3892/or.2023.8502.
- Dutheuil, G. et al. (2025) “Discovery, Optimization, and Preclinical Pharmacology of EP652, a METTL3 Inhibitor with Efficacy in Liquid and Solid Tumor Models,” Journal of Medicinal Chemistry, 68(3), pp. 2981–3003. Available at: https://doi.org/10.1021/acs.jmedchem.4c02225.
- Very recently, an analogous approach was reported: Li, Z. et al. (2024) “A Stapled Peptide Inhibitor Targeting the Binding Interface of N6‐Adenosine‐Methyltransferase Subunits METTL3 and METTL14 for Cancer Therapy,” Angewandte Chemie International Edition, 63(24), p. e202402611. Available at: https://doi.org/10.1002/anie.202402611.
- Work performed with Cresset Discovery: https://www.cresset-group.com/discovery/why-cresset-discovery
- Moroz‐Omori, E. v. et al. (2021) “METTL3 Inhibitors for Epitranscriptomic Modulation of Cellular Processes,” ChemMedChem, 16(19), pp. 3035–3043. Available at: https://doi.org/10.1002/cmdc.202100291.
- Yankova, E. et al. (2021) “Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia,” Nature, 593(7860), pp. 597–601. Available at: https://doi.org/10.1038/s41586-021-03536-w.
- Guirguis, A.A. et al. (2023) “Inhibition of METTL3 Results in a Cell-Intrinsic Interferon Response That Enhances Antitumor Immunity,” Cancer Discovery, 13(10), pp. 2228–2247. Available at: https://doi.org/10.1158/2159-8290.CD-23-0007.
- R. Miller, R. et al. (2020) “Integrating the Impact of Lipophilicity on Potency and Pharmacokinetic Parameters Enables the Use of Diverse Chemical Space during Small Molecule Drug Optimization,” Journal of Medicinal Chemistry, 63(21), pp. 12156–12170. Available at: https://doi.org/10.1021/acs.jmedchem.9b01813.
- Pennington, L.D. et al. (2024) “Property-Based Drug Design Merits a Nobel Prize,” Journal of Medicinal Chemistry, 67(14), pp. 11452–11458. Available at: https://doi.org/10.1021/acs.jmedchem.4c01345.
- Spark. Cresset®: Litlington, Cambridgeshire, UK, https://www.cresset-group.com/software/spark
- Cheeseright, T. et al. (2006) “Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation,” Journal of Chemical Information and Modeling, 46(2), pp. 665–676. Available at: https://doi.org/10.1021/ci050357s.
- Lead Finder; BioMolTech®: Toronto, Ontario, Canada,https://www.cresset-group.com/software/leadfinder.
- Bauer, M.R. and Mackey, M.D. (2019) “Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes,” Journal of Medicinal Chemistry, 62(6), pp. 3036–3050. Available at: https://doi.org/10.1021/acs.jmedchem.8b01925.
- Flare. Cresset®: Litlington, Cambridgeshire, UK, https://www.cresset-group.com/flare.
- Wang, P., Doxtader, K.A. and Nam, Y. (2016) “Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases,” Molecular Cell, 63(2), pp. 306–317. Available at: https://doi.org/10.1016/j.molcel.2016.05.041.


