
摘要:有很多原因需要进行骨架跃迁计算:避开专利保护的分子骨架或侧链以获得自己的专利化合物;扩充专利保护范围;跟进新领域;设计新分子、突破原有化合物缺陷(比如原有化合物代谢不稳定)而保留活性。本教程以EGFR激酶抑制剂为例说明骨架跃迁的计算流程:演示了如何利用SPARK对选定的骨架进行生物等排替换,生成的化合物具有与原型化合物相似的形状与静电但是具有新的骨架,这种新骨架的化合物具有很高的概率保留有原型化合物的生物学活性。
肖高铿 2016/03/31
一. 背景
1. SPARK骨架跃迁可以解决的技术问题
有很多原因需要进行骨架跃迁计算:避开专利保护的分子骨架或侧链以获得自己的专利化合物;扩充专利保护范围;跟进新领域;设计新分子、突破原有化合物缺陷(比如原有化合物代谢不稳定)而保留活性。
2. SPARK进行骨架跃迁的技术原理
形状与静电是与配体与受体相互作用直接相关的两个分子性质,如果一个化合物与受体在形状与静电上互补,那么这个化合物就自然而然地成为该受体的配体。如果两个化合物在形状与静电上相似,那么这两个化合物可能可以结合到同一个蛋白的结合口袋。图1展示了化合物的3D结构(A)、形状(B)与静电(C)之间的关系。但是,静电场信息太丰富而计算量太大,因此我们采用场点(E)来代替静电场与形状场:将静电浓缩至其极值点来表示在空间中的分布。静电场点相似的化合物其静电相似。SPAKR通过比较分子形状、静电场点来比较化合物之间在形状与静电的相似性。
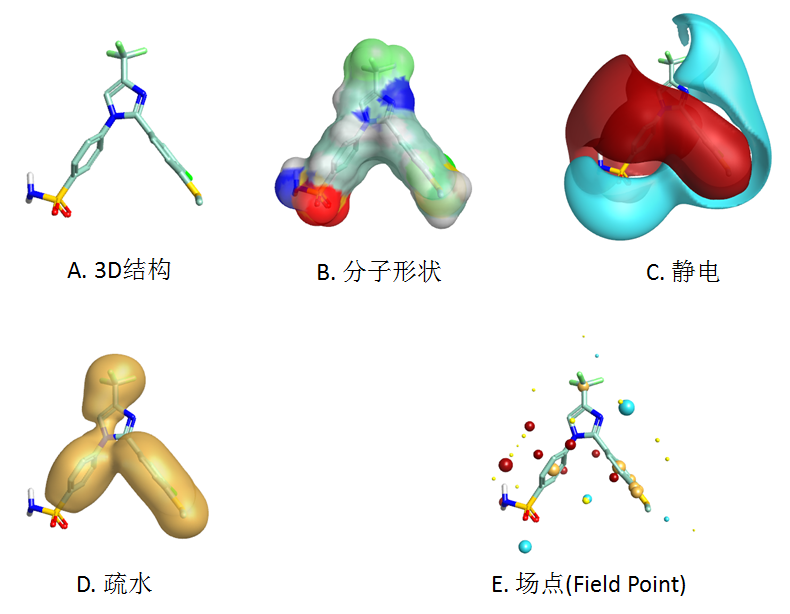
图1. 分子形状与静电以及场点
静电场点与药效团概念很相似,但信息量要比药效团大,比如在药效团概念里苯环只是芳香性疏水中心,而在场点里,除了疏水性之外、还有电荷分布的信息。如图2所示,不同取代基的苯环,其静电场是不同的,尽管药效团描述一样。
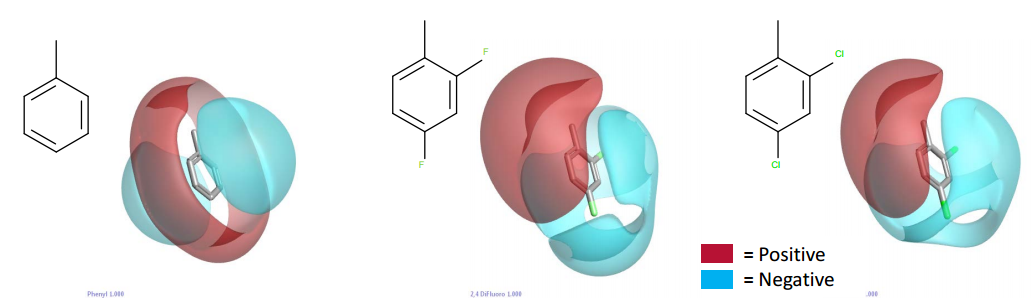
图2. 不同取代基苯环的静电场
SPARK就是利用形状与静电相似性进行骨架跃迁:将骨架替换,而保留形状与静电相似即可达到骨架替换而尽可能保留活性的目的。SPARK可以自动搜索数据库对起始化合物进行结构替换并生成新化合物、将新化合物与起始化合物进行2D相似性比较、分子形状与静电比较、油水分配系数、水溶性等类药性质计算,最后获得合成可行性高、类药性好的全新化合物。
3.SPAKR教程介绍
本教程以EGFR激酶抑制剂为例说明骨架跃迁的计算流程:演示了如何利用SPARK对选定的骨架进行生物等排替换,生成的化合物具有与原型化合物相似的形状与静电但是具有新的骨架,这种新骨架的化合物具有很高的概率保留有原型化合物的生物学活性。
4.SPARK案例
Tuyishime, M., et al. (2016). “Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement.” Bioorg Med Chem Lett 26(1): 228-234. DOI:10.1016/j.bmcl.2015.10.080
Tuyishime, M., et al. (2014). “Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement.” Bioorg Med Chem Lett 24(23): 5439-5445. DOI: 10.1016/j.bmcl.2014.10.027 http://www.molcalx.com.cn/cresset_case_study_04
二. 准备工作
1. 使用的软件
SPARK
2. 跃迁的初始化合物
我们以一个EGFR激酶抑制剂(KJR,IC50=218nM)为例,从其复合物结构4JR3出发,预先下载到硬盘的一个目录里,存为4jr3.pdb。
我们的目的是: 寻找替换高亮部分的骨架。
三. 操作步骤
- 打开Spark,开始一个SPARK项目
- 导入起始的化合物
- 起始物的替换区设定
- 连接位置的原子类型设定
- 片段数据库的选择
- 骨架跃迁(片段替换)
- 结果分析
- 与Forge或Flare联用,预测新设计化合物的活性
双击SPARK桌面图标,启动SPARK,弹出SPARK欢迎界面,点击“New Project”,如图1所示。

图1. Spark欢迎界面
在新弹出的对话框里,选择一个任务,我们选择“Search for bioisosteric replacement”, 如图2所示。
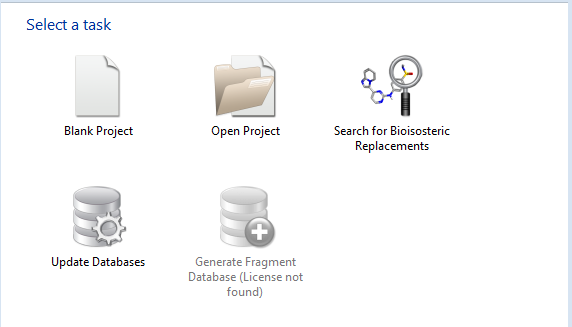
图2. 选择一个任务类型:选择生物等排体置换任务
上一步选中任务类型后,这时弹出新的对话框:Load Starter Molecule,点击PROTEIN按钮,读入预先保存的4jr3.pdb文件,让Spark来推荐质子化状态,如图3所示。
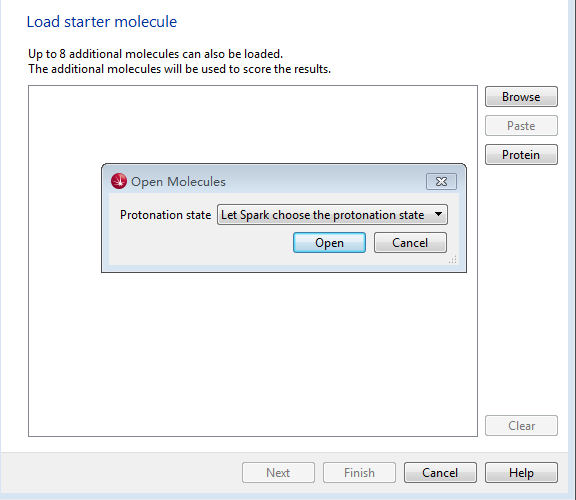
图3. 导入PDB文件4JR3
注:如果小分子起始化合物与其靶点是分离的两个文件,用Browse读入小分子。
点击化学小分子图标,再点击Reference按钮,将复合物结构的化合物设为起始物;再点击Import as protein按钮,导入起始物与蛋白结构,如图4所示。
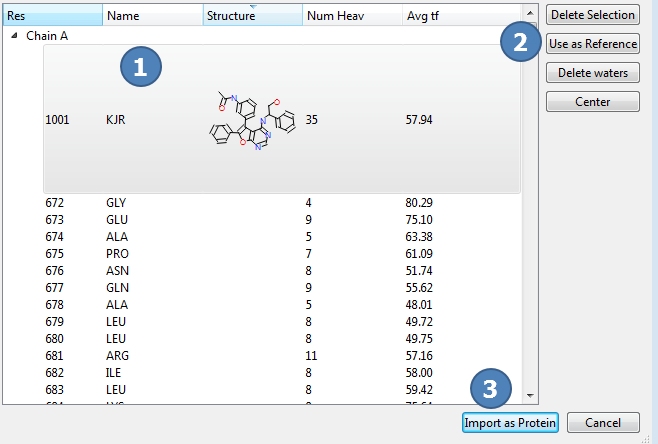
图4.设定起始化合物与蛋白
点击下一步,可以对起始化合物进行确认与编辑,以确保结构正确,如图5所示。如果确认没有问题,点击下一步按钮开始设定起始物的替换区。
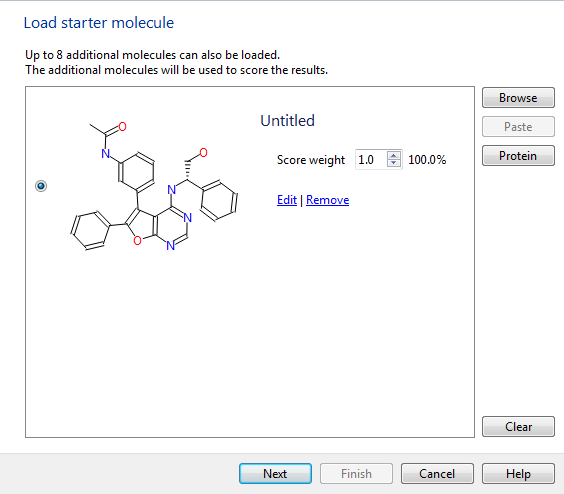
图5.起始化合物的确认与编辑
用按住鼠标左键,将需要进行结构替换的部分圈住,如图6的阴影部分。
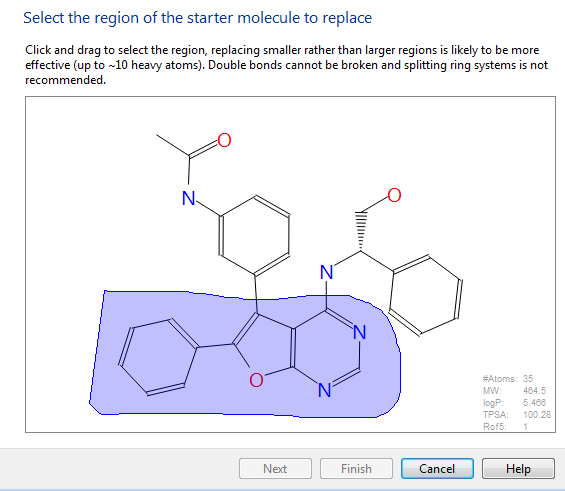
图6.起始化合物的替换区确认
现在SPAKR告诉你起始物需要通过两个连接位点与数据库片段进行连接替换,需要对连接位点的原子类型进行选择定义,主要从合成可行性角度考虑。
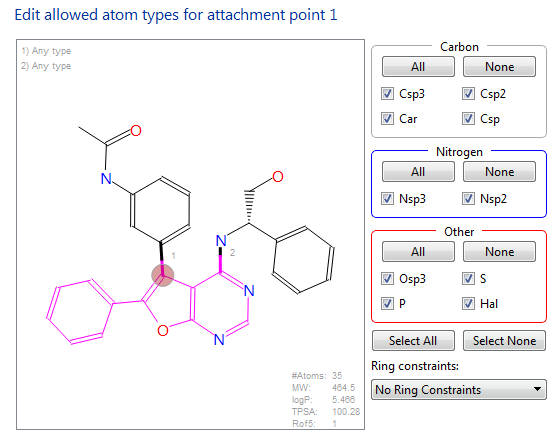
图7.连接位点的原子类型选择
SPARK提供了很多的化合物片段,这些片段都来源于现有的化合物,可以选择性的使用一个或多个片段库,见图8
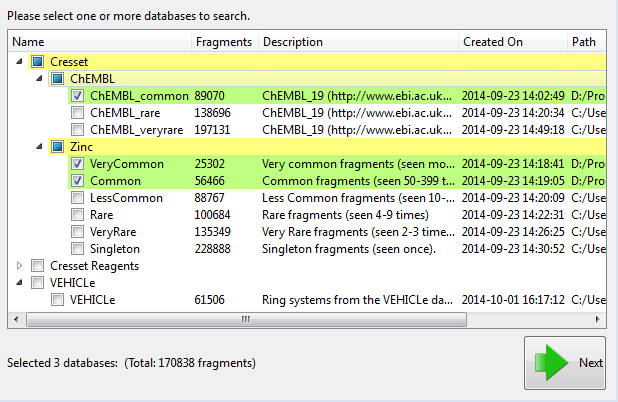
图8.SPARK化合物片段库
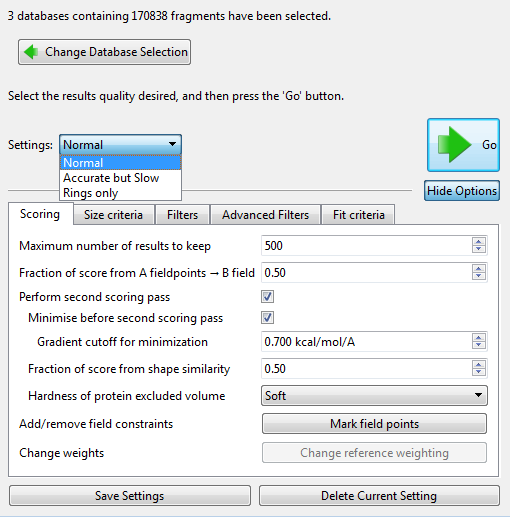
图9.SPARK进行片段搜索时支持多种策略,可以调节各种性质过滤与打分权重
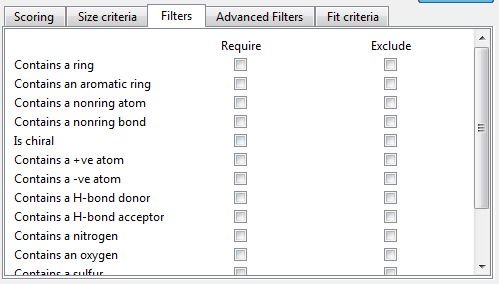
图10.SPARK的过滤条件
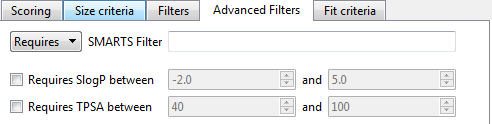
图11.SPARK的溶解度、油水分配系数过滤条件
参数设定好之后,点击Go即开始数据库搜索,自动生成新的化合物
观察新化合物与起始化合物的Field与Shape相似性;
SPAKR提供雷达图等方便的进行多目标分析;
简单的方法是将SPARK的结果用Forge或FLARE读入,再进行计算。比如,打开Forge>新建项目>Fit Molecules to an Activity Model,提示读入模型与带预测化合物,Forge会进一步对化合物进行构象搜索,分子叠合,预测活性,并给出预测的可靠度。
更简单的方式是使用SPARK的send to功能,将结果发送给Flare或Forge进行一下步的活性评估,如图12所示。
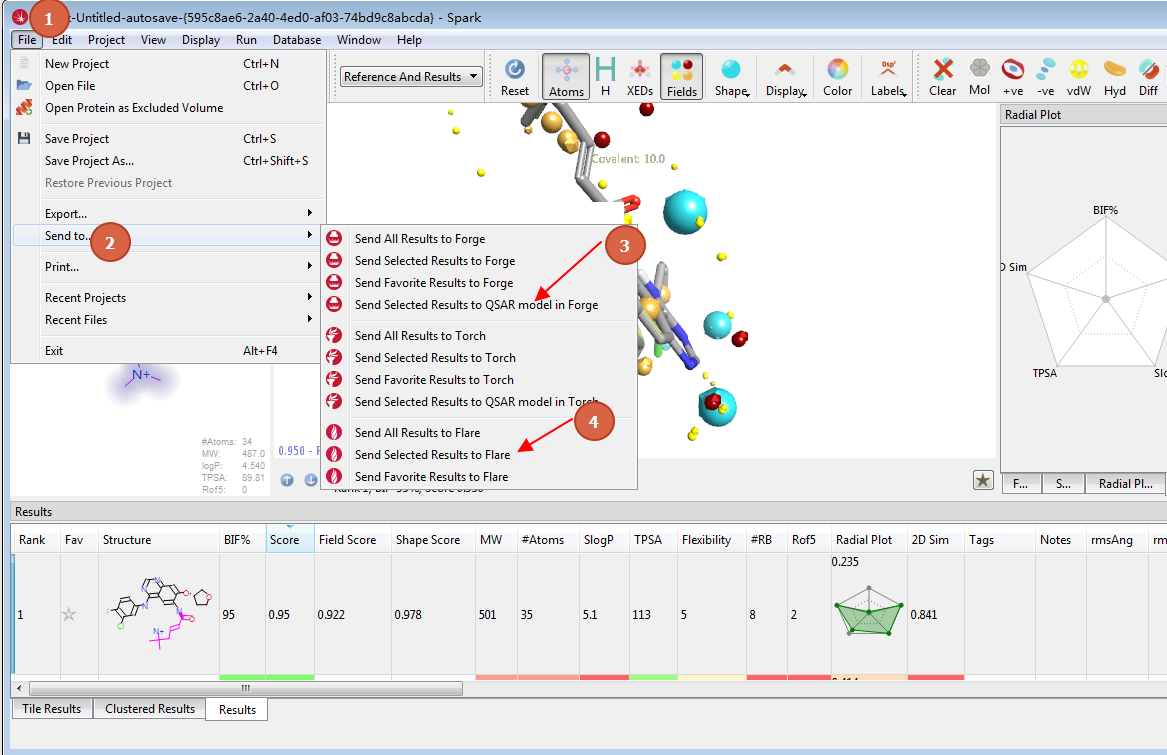
图12.SPARK提供了FLARE与Forge的接口
四. 视频教程
五. 从这里开始还可以做什么
1,分子对接,进一步评估推荐的化合物;
2,用Forge建立的3D-QSAR模型预测化合物的活性;
六. 联系我们获取试用
电邮:info@molcalx.com
电话:020-38261356
单位:广州市墨灵格信息科技有限公司
网站:http://www.molcalx.com.cn
软件下载: http://www.cresset-group.com
试用中文信息提交:


