
摘要:G蛋白偶联受体(GPCR)是细胞表面受体的最大家族,可以说是最重要的药物靶标家族。随着X-射线晶体学和冷冻电子显微镜技术的突破,自2007年以来已公开报道了300多种GPCR-配体复合物结构,涵盖了约60种独特的GPCR。如此丰富的结构信息无疑有助于靶向GPCR开展基于结构的药物设计。在本文中,王任小课题组开发出了一种基于片段的计算方法来设计新型GPCR配体。首先提取GPCR和配体之间的结合界面上的特征相互作用模式(CIP)。 CIP用来搜索源自GPCR配体的化学片段,而这些片段与GPCR形成相似相互作用模式所必需的。然后,使用AutoT&T2软件将选定的化学片段组装成完整的分子。在这项工作中,作者选择了β-肾上腺素能受体(β-AR)和毒蕈碱型乙酰胆碱受体(mAChR)作为靶标以验证该方法。根据计算方法推荐的设计,作者购买了63个化合物的样品,并在基于细胞的功能测试中进行了验证。最后识别了15个β2-AR拮抗剂和22个mAChR M1拮抗剂。还进行了分子动力学模拟和结合自由能分析,以探索这些活性化合物与其靶标GPCR之间的关键相互作用(例如氢键和π-π相互作用)。总而言之,本文提出了一种基于相关3D结构信息进行从头设计GPCR配体的有用方法。

编译:肖高铿
原文:1. Li Y, Sun Y, Song Y, et al. Fragment-Based Computational Method for Designing GPCR Ligands. J Chem Inf Model. 2019;0(0):acs.jcim.9b00699. doi:10.1021/acs.jcim.9b00699
前言
GPCR,也称为七次跨膜受体,是细胞表面受体的最大蛋白质家族。 与细胞外刺激结合后,GPCR激活细胞内信号转导途径,最终调节细胞状态。GPCR可以与各种配体结合,包括光敏化合物、气味、信息素、激素和神经递质。 GPCR参与了一系列的生理过程和疾病状态。 它是制药行业最重要的药物靶标家族,这一事实可以证明,目前市场上超过30%的处方药物均以GPCR为靶标。
由于膜蛋白的溶解度低和稳定性差,解析GPCR的晶体结构一直是具有挑战性的,直到2000年,哺乳动物GPCR的第一个晶体结构牛视紫红质被解析。在很长一段时间内,它被用作构建视紫红质样GPCR同源模型的标准模板。尽管同源建模广泛用于对接方法进行GPCR的配体虚拟筛选,但由于GPCR结构数量有限,GPCR配体的发现仍依赖于高通量筛选或基于配体的虚拟筛选方法。随着人类GPCR的第一个晶体结构β2-AR于2007年发布,GPCR晶体学以及最近的cryo-EM已经取得了重大突破。已经报道了300多种具有激动剂、拮抗剂或反向激动剂结合的GPCR结构,涵盖了来自A、B、C和F类的约60种独特的GPCR。GPCR结构的丰富性为基于结构的药物设计提供了新的机会。
在2015年,Fidom K.等人。报道了一种新的基于晶体结构片段的GPCR药效团方法。在该方法中,使用具有配体结合的62个A类GPCR结构来提取结构片段,即残基-片段相互作用对。片段可以叠合到所选A类GPCR结构模板的7TM骨架上。然后选择代表性片段以产生药效团模型。以组胺受体H1和H3为例,通过回顾性虚拟筛选实验阐明了药效团模型的良好性能。同样在2015年,Kooistra A.J.等人发表了一种方法,该方法使用配体结合的β-AR晶体结构的蛋白质-配体相互作用指纹(IFP)来区分已知β-AR部分/完全激动剂、拮抗剂/反向激动剂和理化性质相似的decoy。该方法在选择性虚拟筛选具有特定功能作用的配体方面显示出优势。结合分子对接的IFP打分也被证实可以改善其他GPCR配体的虚拟筛选富集能力。但是,基于片段的药效团方法和基于IFP的方法都有一定的局限性。前者依赖于结构保守的7TM腔,目前仅限于A类GPCR,因为不同GPCR类之间的药效团元素不易比较。后者在准确反映蛋白质-配体相互作用方面存在问题,因为在将3D相互作用转换为2D指纹时,一些详细信息(例如两个相互作用的原子之间的距离)被遗弃了。此外,这两种方法主要应用于虚拟筛选,而不是从头进行配体设计。
在我们先前对蛋白质-蛋白质相互作用的研究中,提出了蛋白质-蛋白质结合界面上的特征相互作用模式(CIP)的概念,其定义为一种蛋白质链中的一个残基与另一个蛋白一条链中的三个最近残基之间的相互作用。对蛋白质-蛋白质复合物的非冗余数据集的分析表明,CIP在相同蛋白质家族的蛋白质-蛋白质结合界面中保守。 CIP被进一步用于设计小分子配体,这些配体被证明可调节Bcl-2抗凋亡蛋白与其结合肽之间的相互作用。在本研究中,我们将CIP概念从蛋白-蛋白结合界面扩展到了蛋白-配体结合界面。更具体地说,是GPCR-配体结合界面,它是由配体的片段和GPCR蛋白上的三个最接近的残基形成的。以这种方式,期望片段和其附近残基之间的相互作用被更准确地描述。在实践中,我们提取了两个GPCR(β-AR和mAChR)的蛋白质-配体复合物上的CIP,并用它们来虚拟筛选可与GPCR蛋白质结合位点中的关键残基结合的片段。此外,通过使用从头设计程序AutoT&T2,将筛选出的片段顺序组装以生成新的分子结构。根据设计的结构,总共购买了63个具有相似骨架的化合物,并在基于细胞的功能测试中进行了验证。确认了其中15个化合物是β2-AR的拮抗剂,证实了其中22个化合物是mAChR M1的拮抗剂。因此,这种新颖的基于片段的设计方法在利用蛋白质-配体相互作用结构信息进行GPCR药物发现中具有巨大的潜力。
方法
1. 总流程
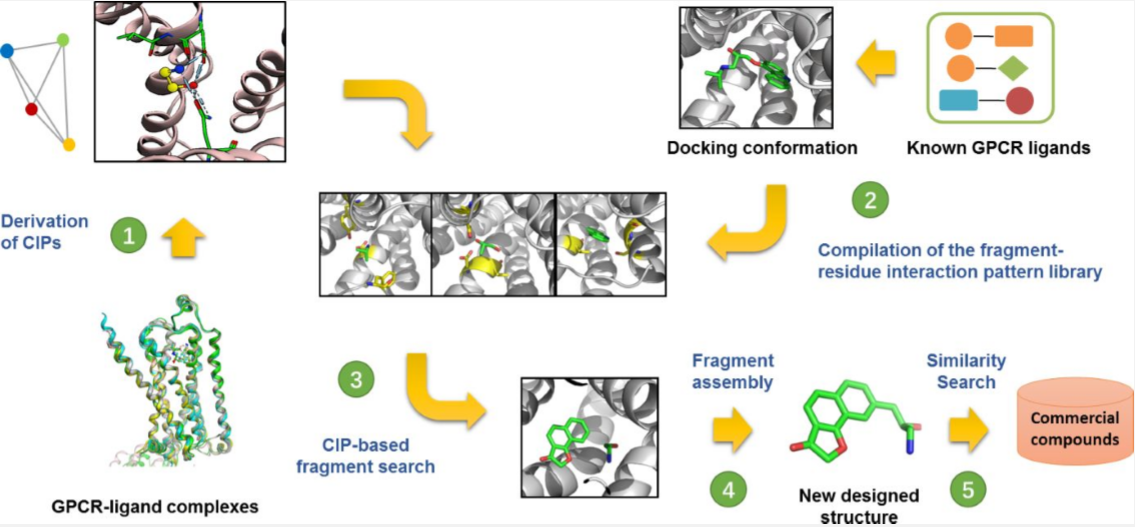
Figure 1. 基于片段的GPCR配体设计的流程图。(1)从GPCR-配体复合物结构生成CIP;(2)通过分子对接从已知的GPCR配体中构建片段-残基相互作用模式库;(3)通过基于CIP的相似度计算进行片段搜索;(4)将搜索到的片段组装起来设计新结构;(5)通过相似性搜索从商业化合物库中获得候选化合物
我们基于片段的GPCR配体设计方法的总体流程如图1所示。第一步,分析GPCR-配体复合物的晶体结构以获得CIP。 然后,基于已知GPCR配体的对接结合模式汇编片段-残基相互作用模式库。之后,通过相似度计算将库的所有相互作用模式与给定的CIP进行比较。 选择这些相似的相互作用模式并保留相应的片段。 随后将结合界面上的相邻片段组装成可与蛋白质结合的小分子配体。 经过一系列分析和过滤后,使用具有代表性骨架的某些结构从商业化合物库中筛选出作为候选化合物。更多详细信息将在以下各节介绍。
2. GPCR数据集的准备
在本研究中,我们选择了两个特定的GPCR(即β-AR和mAChR)作为靶标,它们都是经过验证的药物靶标。完善的生物活性测试方法可用于表征这两个靶标的配体。更重要的是,这两种受体在PDB中有多个与配体形成复合物的晶体结构,这为应用我们的方法提供了基础。这两个GPCR形成的复合物的晶体结构已从PDB下载(截至2016年11月)。 β-AR结构共有30种,包括β1-AR和β2AR,PDB代码为2Y00、2Y01、2Y02、2Y03、2Y04、2YVW、2YCX、2YCY、2YCZ、2RH1、2VT4、3D4S、3NY8、3NY9、3NYA,3P0G、 3PDS、3SN6、3ZPQ、3ZPR、4AMI、4AMJ、4BVN、4LDE、4LEL、4LDO、4QKX、5A8E、5F8U和5JQH。 mAChR有9个结构,涵盖四个亚型(M1,M2,M3和M4),PDB代码为3UON、4DAJ、4MQS、4MQT、4U14、4U15、4U16、5CXV和5DSG。来自PDB的每个复合物结构都被拆分为一个以PDB格式保存的蛋白质分子和一个以MOL2或SDF格式保存的配体分子。蛋白质和配体结构均使用SYBYL软件(8.1版,由Certara Inc.发行)检查并制备。
3. 相互作用模式分析
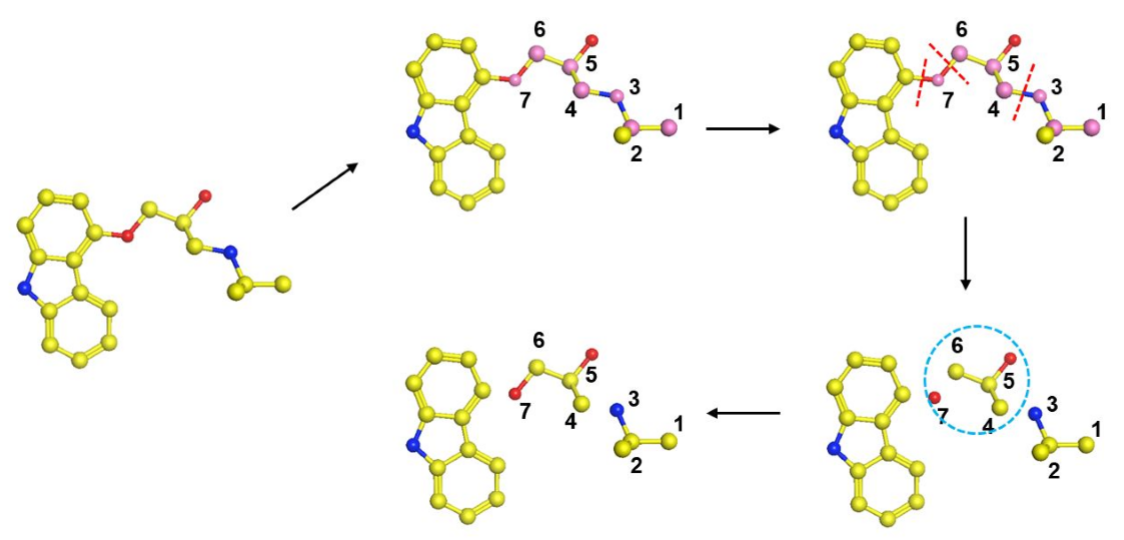
Figure 2.分子碎片化算法
首先使用内部自用的Python脚本将配体分解为片段。 在碎片化过程中应用了一些规则。首先,将单环或稠合/螺环中的原子保留为单独的片段。 丢弃含有四个以上环的片段。 第二,单链片段被定义为具有至少三个连续的无环重原子。 如果存在孤立的原子或基团,则它们会合并到相邻的片段中(参见Figure 2)。

Figure 3.β2-AR与其拮抗剂阿普洛尔(Alprenolol)的复合物晶体结构(PDB:3NYA)中的一个片段与三个最近的氨基酸残基之间的相互作用模式
片段化完成后,分析片段与GPCR之间形成的相互作用模式。相互作用模式定义为包括一个片段及其周围三个最近的残基(图3)。对于一个特定GPCR形成的复合物(例如β-AR或mAChR),记录其所有可能的相互作用模式。可以合理地推测,同一家族蛋白复合物中的CIP应在空间上保守。因此,从片段的相似度和相互作用模式的空间相似性两个方面计算了任意两个相互作用模式的成对相似度值。片段的相似性是根据6个分子描述符来计算的:(1)氢键供体(HBD)的数目;(2)氢键受体的数目;(3)带负电荷的原子数(NEG);(4)带正电的原子数(POS);(5)芳香环(ARO)的数量,和(6)非芳香环(RNG)的数量。然后,通过Tanimoto系数测量最终片段的相似性:
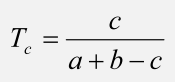
在等式1中,c是两个片段中的公共描述符的数量,a和b分别是两个片段中的总描述符的数量。 如果Tc值低于0.3,则认为片段非常不同。
通过使用我们之前研究中开发的方法,将相互作用模式的空间相似性计算为两个相互作用模式的均方根偏差(RMSD)值。在SI(PART 1)中对此进行了详细地描述。如果一种相互作用模式出现在至少一半的复合物中,并且在不同复合物中的相互作用模式之间的相似性符合定义的标准(Tc大于0.3和RMSD小于1.5Å),则将其选择为蛋白质-配体复合物的CIP。
4.基于CIP的配体设计
一旦确定了给定蛋白质的CIP,我们就可以找到合适的片段与CIP中的残基形成相似的相互作用。只需要计算指定CIP与在其他复杂结构中观察到的相互作用模式之间的相似度计算即可找到这样的片段。原则上,任何蛋白质-配体复合物结构均可用作提供此类相互作用模式的参考。然而,在本研究中为了最大化找到针对给定GPCR靶标的合适片段的机会,我们仅考虑了靶向GPCR的配体来构建该库。从两个公共数据库GPCRSARfari(ftp://ftp.ebi.ac.uk/pub/databases/chembl/GPCRSARfari/)和DrugBank(https://www.drugbank.ca/)下载GPRC配体并进行了处理。将这些对接至包含这两个靶标的蛋白质结构中。一方面,考虑到β-AR或mAChR晶体结构具有相似的结合口袋,将相同的配体对接至相似的结合口袋将导致相似的结合模式。另一方面,同一个配体的碎片化将产生相同的片段集合。因此,可以合理地假设:在不同的蛋白结构中,由相同片段和相同残基对形成的相互作用模式应该具有相似性。因此,只需要一个单一的蛋白质结构即可产生片段-残基相互作用模式。为了验证此方法,我们进行了交叉对接测试。第一步,我们选择了这两个靶标的几种代表性蛋白质结构,它们与数据集中的大多数其他结构包含相同的CIP。然后,将来自我们数据集中晶体结构的所有βAR或mAChR配体对接到这些蛋白质结构上,以分别生成相互作用模式。如果基于来自不同蛋白质结构的对接构象的相互作用模式可以与来自原始晶体结构的CIPs相似,我们认为在这些蛋白质结构上产生的相互作用模式之间的差异可以忽略不计。
根据交叉对接结果,我们最终选择了一些靶标结构(即β2-AR/PDB:2RH1和mAChR/PDB:4U15)用GOLD软件(版本5.2,Cambridge Crystallographic Data Center)生成了它们的结合模式(pose)。 一旦产生了配体对接结合模式(pose),就可以按照上一节中所述的方法获得片段-残基的相互作用模式(请参见SI Part 1中提供的详细信息)。β2-AR和mAChR分别获得400,000和500,000冗余相互作用模式。 这些冗余的相互作用模式的形成主要分为两种情况:一种是相似配体的碎片化生成相同的片段;另一种是在同样的片段生成对最接近的三个残基采用不同的结合构象。
用上一节定义的相似性标准进行基于CIP的筛选。命中的相互作用模式应与残基和可接受的片段匹配,而且化学相似度值应高于0.3。此外,这两个相互作用模式叠合之后的RMSD值应小于1.5Å。 在基于CIP的筛选之后,对于两个β-AR复合物的CIP,分别命中11,993个片段(包含1,663个不重复片段)与8,377个片段(包含1,766个不重复片段)。 mAChR复合结构的三个CIP分别命中7,633个片段(685个不重复片段)、21,765个片段(818个不重复片段)以及8,675个片段(586个不重复片段)。 在结合界面中相邻的片段可以组装成新的结构。
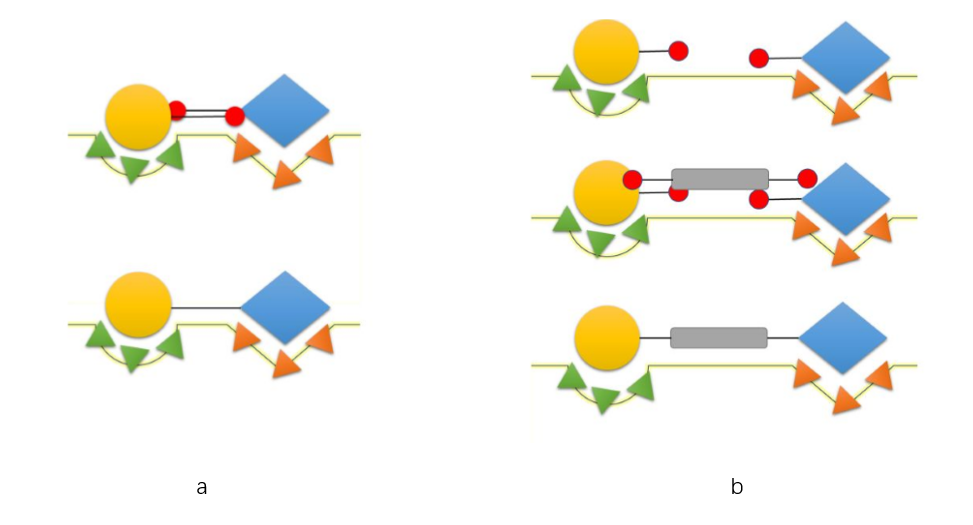
Figure 4. 图解Autot&T2进行基于配体设计。(a)如果两个片段在空间上接近,则沿着匹配的键将适当的子结构截短并组装成新的结构; (b)如果两个片段在空间上相距较远,则首先在它们之间放置一个链接片段(灰色矩形)。 然后重复(a)中的相同操作,将来自这两个片段的子结构以及连接臂顺序组装获得新的结构。新配体设计中使用的子结构用黄色圆圈和蓝色菱形表示。CIP中的残基用小三角形表示。所有的片段都具有一个或两个匹配的键,这些键与一个末端(红色圆圈)相连,末端可以是单个原子(氢或重原子)或短链。
将临近片段组装成新分子用我们课题组开发的AutoT&T2软件(http://www.sioc-ccbg.ac.cn/software/att2)来实现。 AutoT&T2的原理是找到两个分子/结构一对匹配的键,将键两侧的子结构交换生成新的配体。如果在两个筛选到的片段之间有这样一对匹配的键,将合适的子结构剪裁下来并直接连接以形成新的配体(图4a)。 如果两个筛选到的片段相距较远,则首先将连接片段置于它们之间,该片段可选自晶体结构中的参考配体。 然后,使用相同的工作流程将两个片段与链接片段顺序组装在一起(图4b)。
根据AutoT&T2的原理,不同CIP筛选到的片段将按顺序组装起来以生成给定GPCR蛋白的新配体结构。此处,因为除了三个选定的CIP之外还有一个没被占据的子口袋,所以在mAChR的配体设计中还额外多考虑了一个片段集,这将在“结果与讨论”部分中详细讨论。ZINC数据库的“Clean-Fragment”子集包含超过160万个小分子化合物。用Bienfait算法聚类分析后在60%Tanimoto水平得到6,274个代表性片段。这些片段与我们基于CIP搜索获得的片段进行连接。应用相同的对接工作流来生成这些片段的结合构象。
经过基于片段的设计后,AutoT&T2分别为β-AR和mAChR生成了23,000和73,000个原始分子。进行了一系列后处理和分析,以将我们的设计进一步缩小到60个分子:
- (1)“类药性”过滤器:分子量小于600,氢键供体大于5,氢键受体小于10,重原子小于40,环数小于5,可旋转键数小于10和计算的logP值小于6.5。
- (2)由于引入的片段大小的原因,设计的分子可能没有理想的结合模式。因此,用GlideScore将设计的分子重新对接至GPCR蛋白的结合口袋。仅保留对接得分小于-9.0的分子。
- (3)用MOE(2016版)计算每个配体对接模式的蛋白质-配体相互作用指纹相似度。PLIF相似度大于0.60的分子被保留。
- (4)用原子对(atom pairs)和拓扑两面角(Topological torsion)作为聚类分析的分子描述符对剩余分子进行SCA方法聚类,临界值为0.50。
- (5)通过MOE提供的“ rsyn”值来评估设计分子的合成可行性。然后,每个簇(Cluster)选则“ rsyn”值大于70%的化合物作为代表性分子。
5.候选化合物的生物活性测试
将设计出来的分子作为查询结构(query)在商业化合物库eMolecules上搜索相似化合物。结构相似性搜索用Schrödinger软件包中的Canvas模块来进行。使用了三种类型的分子指纹,包括ECFP_4、Molprint2D和拓扑两面角(Topological torsion)。总共购买到了63个候选化合物,其详细结构列在SI中(PART 4)。所有这些化合物都进行了基于细胞的功能测试以检测它们对β2或M1的活性。使用Ultra LNACE试剂盒(Perekin Elmer)进行了基于HTRFFRET的cAMP分析以测试每种化合物对β2受体的激动或拮抗作用,因为β2受体是Gαs偶联的受体。参比激动剂异丙肾上腺素和拮抗剂ICI118551分别用于定义激动剂模式或拮抗剂模式下的最大药效(Emax)。对于mAChRs,由于M1是Gαq偶联的,因此在稳定的M1水母发光蛋白细胞系中进行了基于钙流的水母发光蛋白发光细胞分析。使用参考激动剂乙酰胆碱和拮抗剂pirenzepine-2来分别定义激动剂模式、拮抗剂模式的最大药效。所有化合物均以11点连续剂量的浓度(3倍稀释,最高浓度为100μM)进行测试。GraphPad Prism用来绘制每种化合物的剂量-效应曲线,适应Sigmoidal模型确定各自的EC50或IC50。更多的生物学分析的信息参见在SI(PART 2)。
6.分子动力学模拟与结合自由能计算
选择了几个代表性的苗头化合物用分子动力学模拟和结合自由能分析来探索结构-活性关系。 苗头化合物与靶蛋白的初始结合构象用分子对接程序GOLD(version 5.2)生成。 然后用在线服务器CHARMMGUI(www.charmm-gui.org/?doc=input/membrane)将其浸泡在磷脂双层膜环境中,加上适当数量的抗衡离子和TIP3P水盒。用常规的MD流程来准备GPCR-ligand-POPC复合物结构,其中包括:
- (1) 进行10,000步限制蛋白质和配体的重原子(restraint_wt = 10.0 kcalmol-1Å-2)的能量最小化;
- (2) 解除限制,进行10,000步的能量最小化;
- (3) 在限制蛋白质和配体的重原子(restraint_wt = 10.0 kcalmol-1Å-2)条件下进行500ps的升温:从0K加热到300K;
- (4) 在300K的平衡下解除现在进行500ps模拟;
- (5) 解除限制,在300K温度下进行production模拟持续100ns。
从Production轨迹的最后50ns开始,每10ps采集一个构象,总共采集样5,000个构象。通过MMTSB工具(http://blue11.bch.msu.edu/mmtsb/Main_Page)对这些构象进行了k-means聚类。具有最大成员的簇的质心作为代表性的结合构象。在相同的轨迹上,每500ps采样100个构象。 MM-GB / SA用这些构象进行结合自由能分析。为了使比较具有一致性,将相似的方案对参考化合物ICI118551(β2-AR)与哌仑西平(M1)进行同样的处理。更详细的信息请参见 SI(PART 3)。
结果与讨论
1. 生成β-AR与mAChR的CIP
如图5所示,β-AR受体总共生成了两种CIP。
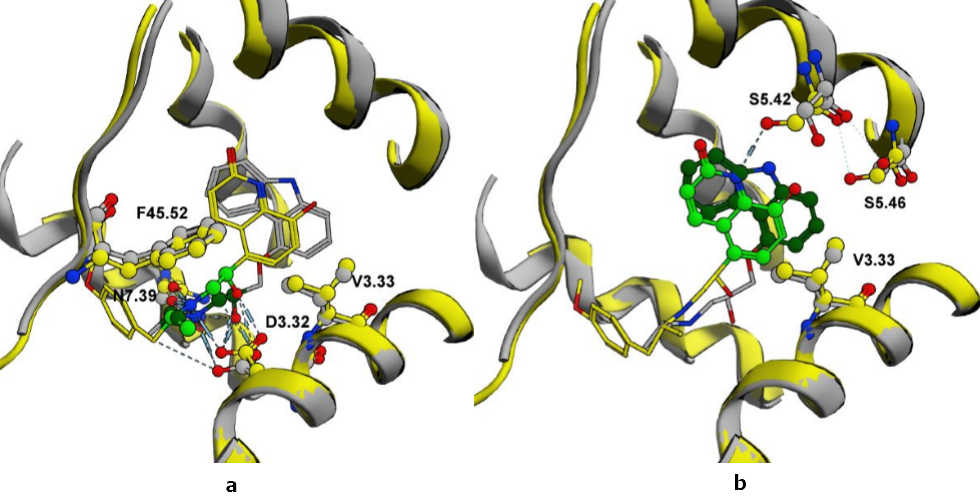
Figure 5. β-AR结合口袋中两个可选择的CIP。在球棒模型中突出显示了形成CIP的氨基酸残基和配体片段。 蛋白质结构的PDB:2RH1(黄色); 2Y02(灰色)。 在这里,两个叠合的结构用于显示CIP的细微差别。
如图6所示, mAChR受体总共生成了3种CIP。
Figure 6. mAChR结合口袋中的三个CIP示意图。在球棒模型中突出显示了形成CIP的氨基酸残基和配体片段。 蛋白质结构的PDB代码:4U15。 CIP未占据的子口袋用虚线圆圈标记。
2. 不同蛋白结构产生的相互作用模式之间的一致性
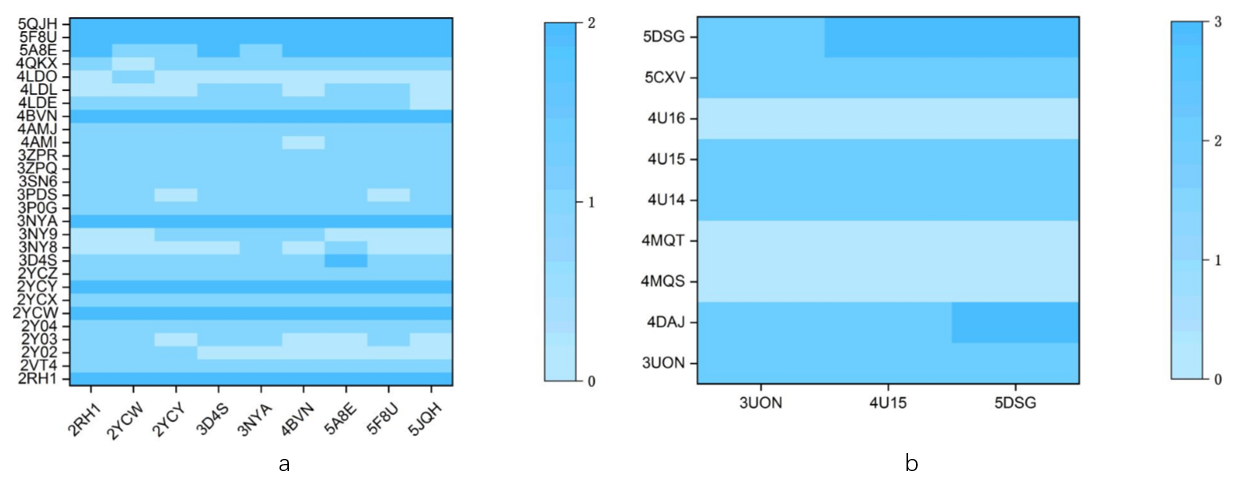
Figure 7. 基于配体对接计算的相互作用模式与在(a)β-AR和(b)mAChR复合物晶体结构中观察到的CIP之间的相似性说明。x轴表示配体对接中使用的蛋白质结构。y轴表示蛋白质-配体复合物的晶体结构。热度图(heat map)按与晶体结构中CIP匹配的相互作用模式的数量着色。
为了准备基于CIP筛选的片段-残基相互作用模式库,我们选择了已知的GPCR配体作为砌块(building-block)片段的来源。注意,为了计算方便,仅使用一个代表性的靶标结构来生成配体的对接构象(pose)。为了验证此方法,我们进行了cross-docking测试。根据这两个靶标的所有蛋白质-配体复合物结构之间的CIP相似性比较(参见SI PART1的Table S1和Table S2)结果,总共选择了9个β-AR结构(PDB 2RH1、2YCW、2YCY、3D4S、3NYA、4BVN、5A8E、5F8U和5JQH)和3个mAChR结构(PDB 3UON、4U15和5DSG)作为代表。这些结构包含与数据集中大多数其他复合物结构相同的CIP。之后,将来自晶体结构的所有30个β-AR配体和9个mAChR配体对接到两个靶标的代表性蛋白质结构中,分别产生所有可能的相互作用模式。我们将基于配体对接构象的相互作用模式与源自其原始晶体结构的CIP进行了比较。可以在热度图(heat map 图7)上看到,不同蛋白质结构的基于配体对接构象的相互作用模式通常与原始晶体结构中获得的CIP一致。换句话说,仅使用所选靶标的一种代表性结构来生成已知GPCR配体的对接构象(pose),然后汇编片段-残基相互作用模式库是合理的。在我们的工作中,对于β-AR和mAChR,分别使用PDB 2RH1和4U15来完成此任务。
3. β-AR与mAChR获得的苗头化合物(hits)
根据方法部分提到的一系列筛选标准,从AutoT&T2生成的23,000和73,000多个原始结构中选择了几种代表性的结构分子。使用这些分子作为查询结构,通过基于指纹的相似度计算从eMolecules中选择并购买了总共63个候选化合物。其中,27个化合物用于β-AR,其余36个化合物用于mAChR。这些化合物都在两个靶标上进行了生物活性测试。它们的化学结构、ID、生物学测试结果列于表S3(见SI,PART4)。
在cAMP分析中测试时,靶向β-AR的27个候选化合物中的15个(〜56%)对β2-AR表现出拮抗活性,IC50值为0.8μM至98μM。根据它们多样的骨架,这些化合物可分为7类(表1)。最大的一类包括八个化合物,它们通常在15个化合物中具有最佳的活性。这些化合物均含有一个羟基,在苯环邻位含有的仲或叔胺基,它们可能与残基D1133.32和N3127.39形成氢键,就像PDB 3NY8晶体结构中所示的已知反向激动剂ICI118551一样。II类化合物具有与I类具有相似的结构特征,但活性低得多。它们均具有相对柔性的末端氨基,这可能影响了氨基与残基D1133.32和N3127.39之间氢键相互作用的形成。其余化合物具有完全不同的结构特征,例如AS-34(图8c)可能与蛋白质具有不同的结合构象有关。
 |
 |
| a | b |
 |
 |
| c | d |
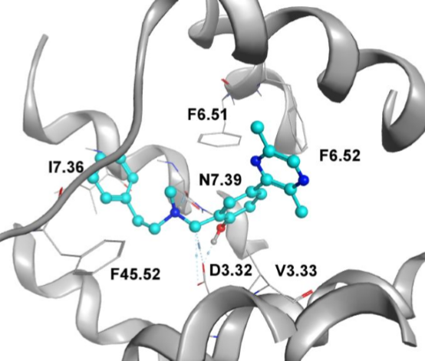
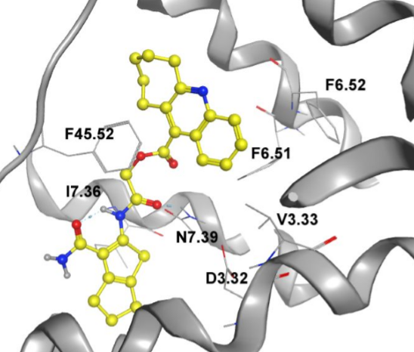
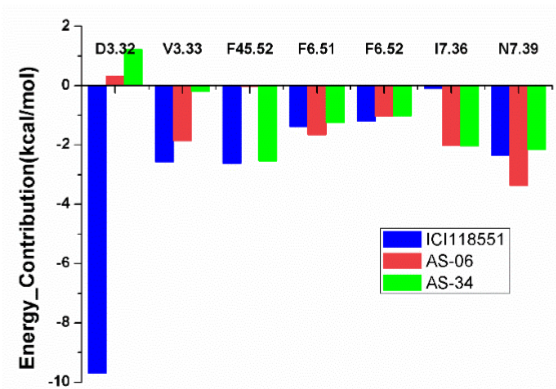
Figure 8. 三种β2-AR配体的预测结合模式(a:ICI118551; b:AS-06; c:AS-34),以及(d)每个残基对结合自由能的贡献。
我们的化合物与现有的肾上腺素能受体拮抗剂相比具有完全不同的骨架,现有的肾上腺素能受体拮抗剂主要含有2-氨基-1-乙醇片段。尽管在我们的测试中,活性最高的化合物AS-06仍比ICI118551(IC50 = 3nM)弱约250倍,但它可作为药物化学hit-to-lead项目的起点。为了解释这种差异并为可能的进一步优化提供指导,用MD模拟和结合自由能预测进行了结构-活性关系分析。图8a-c展示了100ns MD模拟的ICI118551、AS-06和AS-34与β2-AR的预测结合模式。在MD模拟中,ICI118551的胺基和羟基与D1133.32形成稳定的氢键(SI,图S1 a)。在AS-06中,胺基和羟基之间还有一个连接原子,这使质子化的氮更倾向于与N3127.39相互作用(图8b,S1 b)。AS-34可能通过羰基与N3127.39形成氢键(图8c,S1c)。单个残基的结合自由能贡献分析(图8d)表明D1133.32与质子化胺基之间的相互作用可能主导了配体与β2-AR的结合。AS-06和AS-34都从D1133.32获得了非常不利的能量贡献,这可以解释为什么它们与ICI118551之间的有着巨大活性差异。
 |
 |
| a | b |
 |
 |
| c | d |
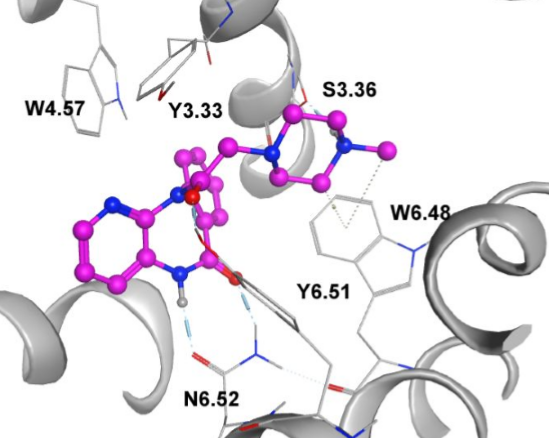
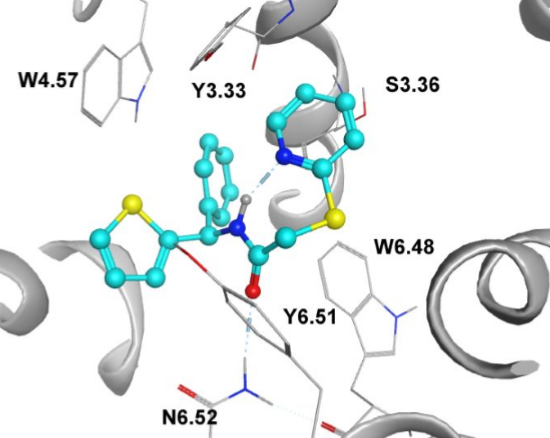
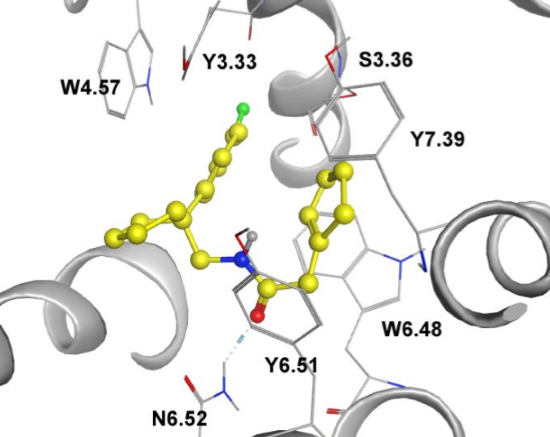
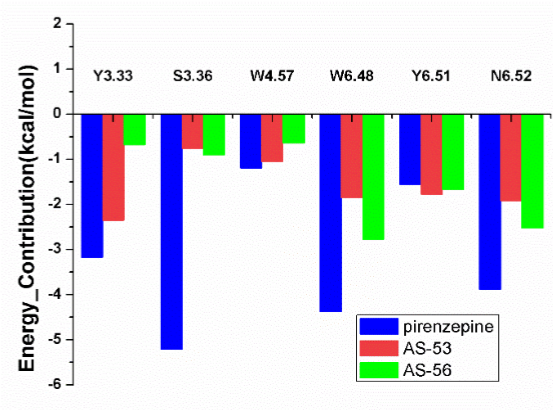
Figure 9. 三种mAChR M1配体的预测结合模式(a:哌仑西平; b:AS-53; c:AS-56),以及(d)每个残基对结合自由能的贡献。
在本研究中,总共购买了36个靶向mAChR的化合物,SI(PART 4)中对这些结构进行了总结。在我们的水母发光蛋白钙发光测试中,其中8个(〜22%)化合物对M1表现出拮抗活性,IC50值在11μM至50μM之间(表2)。我们比较了MD模拟预测的AS-53、AS-56和哌仑西平的结合模式(图9a-c)。MD模拟的氢键分析表明,由于结构中的顺式-酰胺基团使哌仑西平中的酰胺基很可能与N5036.52的侧链形成螯合状氢键(SI Figure S2 a)。我们的苗头化合物AS-53和AS-56仅提供羰基与N5036.52形成氢键,这导致了它们比哌仑西平对结合能量贡献要小(Figure 9d)。此外,哌仑西平具有质子化的哌嗪氮原子,该氮原子可与S1513.36形成极性相互作用。它似乎对结合自由能有着很大的贡献(Figure 9d)。然而,我们所有的化合物都缺乏这样的质子化氮原子,无法与S151形成潜在的相互作用。所有这些结构上的差异都可以解释为什么我们的化合物具有相对较低的生物活性。
有趣的是,最初为靶向β2-AR设计的一些化合物实际上对M1表现出拮抗活性(Table 1)。其中有些甚至比针对M1的化合物活性更好。显然,那些非选择性拮抗化合物都具有一个共同特征:含质子化N原子的烷基链或烷基杂环。该部分可与S1513.36形成相似的极性相互作用,从而有助于M1拮抗剂的功能活性。实际上,许多设计分子确实含有带正电荷的胺基。不幸的是,用相似性搜索的买到的化合物缺乏此结构特征。一种可能的原因是基于指纹的相似度计算具有局限性。在当前的相似度计算方法中,每个定义的分子特征具有相等的权重。因此,如果两个结构具有共同的期望特征,则由于具有更多不同的特征,仍然可能获得小的相似度值。因此,在基于结构的设计中应考虑对官能团特征的重视。一种可行的策略是在相似度计算中增加关键分子特征的权重因子。
我们将所有候选化合物重新对接至β-AR和mAChR靶标结构中以检查它们是否仍能满足所需的CIP。我们发现,所有β-AR候选化合物都能够匹配至少一种所需的CIP。特别是,在15个已确认的β2-AR拮抗剂中,有11个可以满足两种源自晶体结构的CIP(请参见SI PART4的Table S4)。 在36个mAChR候选化合物中,有30种能够匹配至少一种所需的CIP。在已确认的M1 mAChR拮抗剂中,只有AS-60可以满足所有三个所需的CIP(请参见SI,PART4 Table S5)。这些CIP分析结果在某种程度上反映了实验测定的候选化合物活性趋势。具有更多所需CIP的化合物可能是靶向GPCR蛋白的活性配体。 这也提醒我们,在实际实验之前,CIP分析可能有助于排除非活性化合物。
结论
我们利用从GPCR-配体复合物结构得到的CIP开发了基于片段的设计方法。每个CIP由配体上的一个片段和GPCR靶标上的三个最接近的残基来定义。片段和相互作用残基的物理化学性质,以及由片段和相互作用残基的质心表示的四面体叠合的RMSD值来定义的空间相似性,两者都用可用来比较相互作用模式。一旦找到匹配的相互作用模式,结合的片段将被认为适合于该位置。因此,该计算方法可用于筛选片段,并使得随后进行的基于片段的药物设计就像进行湿实验一样。我们的方法已经在两个A类GPCR上进行了实验验证,即β-AR和mAChR。在63个测试过的候选化合物中,共确认了15种β2-AR拮抗剂和22种M1拮抗剂。因此,靶向β-AR和mAChR的化合物的成功率分别为56%和22%。这样的成功率水平代表了传统虚拟筛选工作的显着提高,传统的虚拟筛选工作通常成功达到10%-30%。
通过我们的方法,对GPCR具有已知药理活性的配体可用于衍生片段,以构建片段-残基相互作用模式库。然而,设计的配体不限于在已知的GPCR配体中观察到的结构骨架。 例如,我们获得了7种不同的β2-AR拮抗剂。 其中的一些含有2-(氨基甲基)-苯酚或2-(哌啶-1-基甲基)-苯酚基团,以模仿已知的β2-AR拮抗剂中的2-氨基1-乙醇骨架。其他一些配体也包含完全不同的片段而没有相邻的羟基和氨基。这证明了我们基于片段的方法在为给定的GPCR靶标生成结构多样的设计中的作用。
我们当前的方法也存在局限性。在本研究中发现的活性化合物比已知的拮抗剂弱约100-10,000倍。这有两个原因:首先,当前基于指纹的相似性计算方法无法考虑某些关键结构特征的重要性,例如,在发现M1拮抗剂时忽略了氮杂双环或氮杂三唑基。其次,我们的方法选择与CIP匹配的片段进行配体设计。为了产生片段-残基相互作用而采用的分子对接可能会引入不精确的结构,这会导致将一些对结合亲和力没有重大影响的片段选入最终设计的配体中。应该提到的是,最近机器学习已应用于识别选定靶标的活性专一片段。这一新趋势为进一步优化基于CIP分析的基于片段的设计方法指明了一个有希望的方向。
文献
略,请阅读原文。
推荐类似的文献或算例
虽然作者没有提供CIP识别、CIP虚拟筛选、分子碎片化的源代码或工具,但是可以借鉴其思想用类似的软件实现。下面的两个例子可供参考,请点击标题阅读原文。
- 基于药效团片段的虚拟筛选:发现高效先导化合物的强有力方法
- Ligandscout教程 | 基于片段的虚拟筛选联用AutoT&T进行从头设计
欧莱雅公司的研究人员对已经批准上市的美容用分子进行碎片化、官能团化与重新组合等一系列处理,构建了适于虚拟筛选的虚拟库。对该虚拟库进行了基于药效团片段的虚拟筛选以及三个月的优化阶段后,发现了一个各种性质都理想的先导物,其有望在美容应用领域里作为皮肤护理的候选分子。本项目的成功为其它美容靶点的先导物发现铺平了道路。
本文演示了如何将Ligandscout的基于片段虚拟筛选与AutoT&T 2联合使用,实现化合物的从头设计。首先用Ligandscout进行基于结构的药效团模型识别;再将选定的药效团特征设置为optional进行基于片段的虚拟筛选;最后用AutoT&T 2将命中的片段链接到先导化合物的保留片段上生成全新的化合物。实际上,CIP就是个药效团片段。


