
数据库挖掘的启示:不同寻常的蛋白-配体相互作用有多重要?
摘要:我们提出了一种新方法,可以从PDB的挖掘中获得蛋白-配体原子对的相互作用倾向。为了确保得到可靠的统计,我们使用视线接触过滤器,并通过基于蛋白−配体结合位点中原子类型的暴露表面积的统计学空模型对观察到的命中频率进行归一化。这使得我们能够研究在蛋白-配体复合物中什么样的分子间相互作用及其几何结构会被我们频繁而不止于随机地发现。本研究聚焦于一些被认为是有利的、不同寻常的相互作用上,包括卤素和硫原子的σ-空穴键(bonding of σ-hole)、以氟为受体的弱氢键以及不同类型的偶极相互作用。我们的结果证实并挑战了关于这些相互作用的一些常见假设,并强调了在基于结构的药物设计中尚未被探索的其他接触类型。
原文: Kuhn, B.; Gilberg, E.; Taylor, R.; Cole, J.; Korb, O. How Significant Are Unusual Protein–Ligand Interactions? Insights from Database Mining. J. Med. Chem. 2019, 62 (22), 10441–10455. https://doi.org/10.1021/acs.jmedchem.9b01545.
编译:肖高铿/2021-05-01
前言
在基于结构的药物设计中,通过分析有利和不利的蛋白-配体相互作用来理性解释化合物的结构-活性关系的局部相互作用模型非常普遍,尽管存在局限性,但它在药物发现过程起到增值作用[1]。在过去十年中,一直被认为是有利的、且在药物设计过程中原则上应考虑的相互作用类型的数量大幅增加。其中包括弱氢键,它的氢键供体或受体不是氧或氮原子,而是极化的CH基团[2]、与碳成键的卤素[3]、或电子π系统[4,5]。一系列实验和理论模型研究不仅证明了重卤素与路易斯碱之间的σ-空穴键(σ-hole bonding)的有利贡献[6−8],而且还声称氟[9]、硫[10−13]、氮[14]或磷[15,16]可以在某些情况下参与这种类型的相互作用。人们强调了多极相互作用的重要性[17],除了阳离子−π相互作用外,阴离子−π相互作用[18−20]也有其支持者。所有这些潜在有利的新相互作用类型,在本文中称之为“不寻常的”相互作用。人们可能会好奇,在存在多种相互作用选项的蛋白-配体结合位点环境中,哪些相互类型真正地具有竞争优势呢?
蛋白-配体复合物晶体结构数据库(例如蛋白质数据库,PDB[12])可以极大地帮助回答这个问题。挖掘它们丰富的实验内容提供了对分子间相互作用偏好的见解,并允许对其与分子识别的相关性进行统计评估。有许多研究分析了PDB中不同类型的相互作用模式(interaction motif)[22-30]或将其内容用于推导基于知识的打分函数[31-35]。一个关键且经常被忽视的问题是如何对特定接触类型观测到的次数进行归一化以便对推导出的统计学数据进行的解释是有意义的。泰勒(Taylor)[36]最近引入了一个统计框架,其使用了空假设,即与某原子类型发生相互作用的概率仅取决于该类型原子所贡献的分子表面积比例。然后,将之用于分析小分子剑桥结构数据库(Cambridge Structural Database, CSD[37])以推导分子间相互作用偏好。在我们的研究中,将泰勒的概念用于蛋白-配体结合位点,并用一个角度分量加以扩展,以解释诸如氢键或σ-空穴键等等具有高度方向性的相互作用类型。由此,我们可以探索一个给定的相互作用类型在PDB中的出现(频率)可以达到何种程度,以及该相互作用类型是否比起预期的随机更加频繁地出现。如果是这种情况,我们认为这种相互作用类型在蛋白-配体识别中是有利的,并具有竞争优势。
我们的目的是更清楚地了解一些不寻常的分子间相互作用的意义,比如σ-空穴键、弱氢键以及偶极等等相互作用。 我们通过识别卤素和硫原子以及诸如腈和磺酰基之类具有显著局部偶极矩的基团的优选蛋白环境来做到这一点。这些官能团通常用于结构指导的药物化学优化,增加对它们优选的相互作用对象和几何结构的了解将使得优化过程更加高效。
结果与讨论
表1. 本研究涉及的配体官能团SMARTS模式

a. SMARTS index的第一个数字(原子的编号从0开始)指示了用来计算SMARTS模式中配体的原子(以粗体字显示)到相互作用的蛋白原子之间的距离(比如图1中,L1用来计算L1-P1之间的距离)。第2个数字指示了用来计算相互作用角度的配体原子(比如图1中,L2用来计算L2-L1-P1角度)。
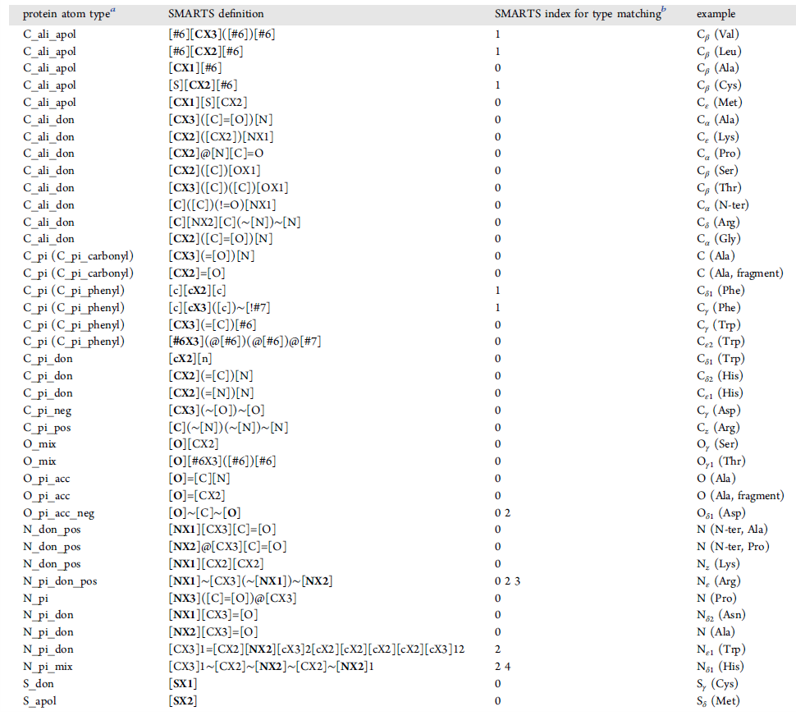
表1列出了我们分析过的蛋白环境下的配体官能团。我们将作为相互作用对象的蛋白原子根据其元素、氢键特征(don, acc,mix,apol)、原子电荷(pos,neg)和电子系统(ali,pi)的不同,分为16种不同的类型(表2)。此外,相互作用类型X是指结合位点上其它HET基团的接触原子,我们还对蛋白原子类型的组合(称为超集)进行了分析,如表S3所定义。
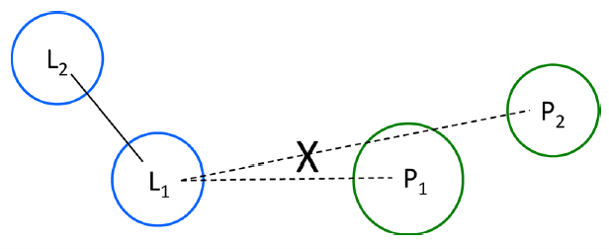
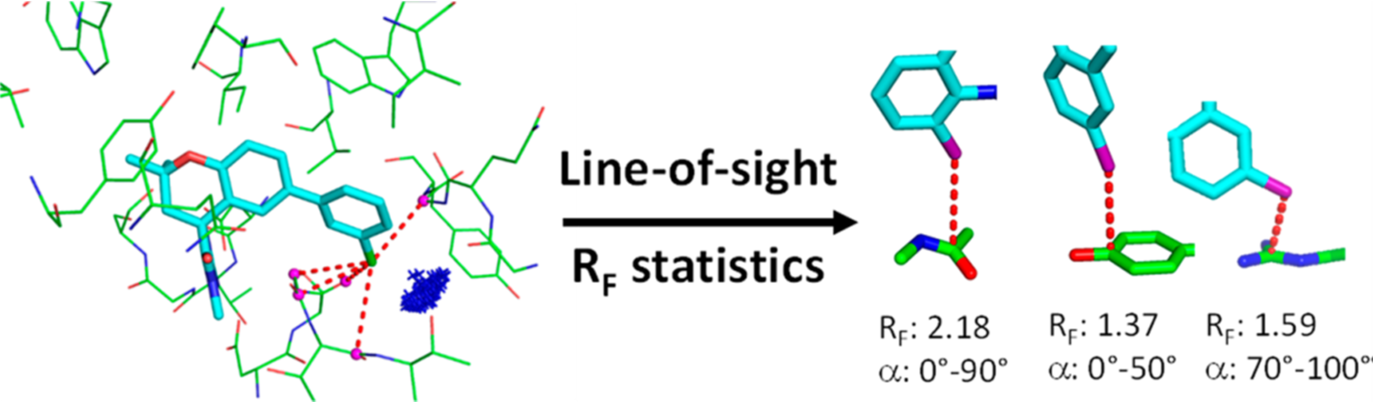
图1. 本研究所用的视线概念(Line-of-sight concept)示意图。虚线表示配体原子Li与蛋白原子Pi之间的非键接触,实线为共价键。仅计数配体原子L1和蛋白原子P1之间的分子间接触。如果原子P1的范德华球面与连接L1和P2中心的线相交时,则删除L1… P2接触。如果配体和蛋白原子之间的距离小于其范德华半径[38]之和加0.5 ,则将相互作用计入在内。
本方法有几个重要的特点值得一提。首先,我们的分析仅包含“视线”(Line-of-sight, LoS)相互作用(图1),从而避免了计数来自旁观原子的二次接触,后者仅由于与形成一级接触的原子通过共价连接而存在。我们认为,不应将二级接触包括在内,我们希望仅聚焦于LoS相互作用,以使得相互作用统计更加清晰。
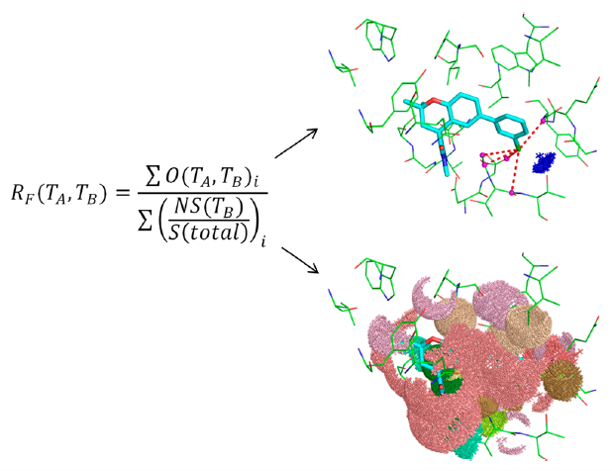
图2. 用于计算RF相互作用倾向的基于表面的统计方法示意图(eq 2;详细内容见实验部分)。右上角显示了配体查询原子在蛋白结合位点中的LoS接触(虚线),蓝色表面是配体原子的溶剂可及表面区,用水映射表3将该区域转换为显式的水接触。图片底部显示了暴露在结合位点中的表面,在eq2的分母中会用到,根据蛋白原子类型TB进行着色,配体的溶剂可及表面积单独用一种颜色着色。
其次,根据Taylor[36]对小分子CSD数据的研究,建立了一个可靠的统计模型来推导蛋白-配体相互作用的倾向。如图2所示以及“实验”部分的详细说明,本研究将在PDB中观察到的给定类型的LoS接触数量与从表面积考虑得出的预期数量进行了比较。具体而言,配体原子与给定类型的蛋白原子形成接触的概率是该类型原子的暴露表面积与蛋白-配体结合位点中总暴露表面积的比值。频数比值(Ratio of frequencies,RF)可以使用方程(2)计算,统计误差用自举法计算。我们进一步对之前方法进行了改进,这使得我们能够用角度约束的情况下进行RF分析。这一点很重要,因为它捕获了强方向性相互作用(例如σ-空穴键)的几何偏好。RF大于1的相互作用要比随机的更加频繁被观察到,我们认为这些相互作用,在确认过的优选几何结构范围内,在蛋白结合位点内是有利并具竞争优势的。显然,相互作用的方向性越强,配体以形成相互作用的方式结合的可能性就越小。但是,如果有可能的话,定向相互作用可能会具有非常强的竞争优势。
表3. 蛋白原子类型及其SMARTS定义、用于原子类型匹配的SMARTS模式索引
最后,与结合位点中的水分子接触更加难以进行统计学评估,因为在蛋白质晶体结构中被明确地解释的水只是其中的一部分。 为了让我们的理论框架也能应用于水接触,我们使用了一种变通方法,首先确定结合位点中配体查询原子的溶剂可及表面积(SASA),并用映射表(表3;有关详细信息,请参阅实验部分)得到一个期望的水接触数量。对于涉及水的相互作用分析,我们将在整个文本中使用“watmap”这个原子类型。
表3. 水映射表将配体原子(删除所有水分子)溶剂可及表面积(SASA)与期望的显式水接触数关联起来
卤素的相互作用

图3. 配体中与芳香碳键连的氯原子的RF相互作用统计图解。x轴表示表2和S3中列出的不同的蛋白接触原子类型,y轴上的柱状图高度为RF值以及自举法推导的误差条。RF=1的水平线指示随机期望的RF值。与水的相互作用统计(原子类型“watmap”)用黑色虚线矩形标记,在95%置信区间内RF大于1的蛋白原子类型用绿色高亮显示。额外的角度约束未加以考虑。
统计分析得出的相互作用倾向性用RF值表示,图3用柱状图描述了连接到芳环上的配体氯原子统计分析结果。图表左侧展示了与氯原子发生相互作用的蛋白原子类型,它们具有统计学意义上的显著性(RF值大于1),这表明在PDB中发现这些蛋白原子类型的频率不止于期望的偶然发现。这样的相互作用发生于极化的CH基团(C_ali_don,RF=1.23±0.07),π系统的碳原子(C_pi,RF = 1.21±0.05)和非极性原子的超集(apolar,RF = 1.02±0.02)。误差线相对较小,反映了配体氯原子在蛋白结合位点中的良好覆盖。氯原子与疏水性或弱极性蛋白碳原子的相互作用占优势,水(watmap)或极性超集(don,acc,pos,neg)相对较低的RF值(小于1),这突显了氯原子的强疏水性特征。
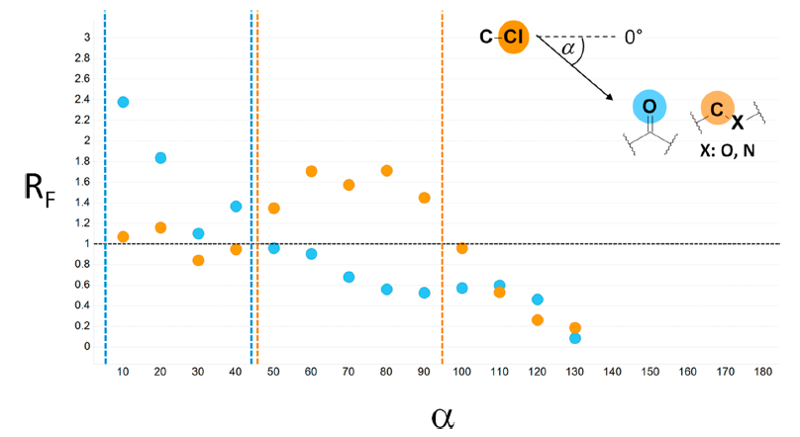
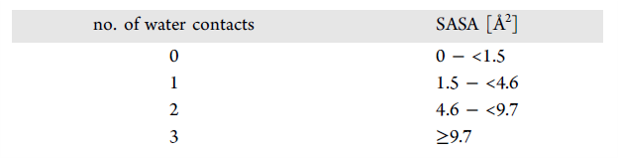
图4. 与芳香碳原子键连的配体氯原子与两种不同类型蛋白原子O_pi_acc (蓝色) 及 C_ali_don (橙色)相互作用的角度RF统计。x-轴为增量步长为10°的α角,该α角定义为C-Cl键延长方向角度偏差。RF=1的水平线随机期望的RF值。竖直虚线定义了与O_pi_acc(蓝色)以及C_ali_don (橙色)相互作用有利的α角度范围。
如果达到特定的距离和角度阈值,则可以推测氯原子与更强极性的蛋白之间的相互作用(例如,证据充分的与孤对电子供体之间的 σ-空穴键)被认为是有利的[8]。虽然我们在与角度无关的图3中与羰基氧原子(O_pi_acc)间的相互作用没有看到高的RF值,但对含有相互作用角度的数据进行统计分析时,所得的图看起来有所不同。图4的蓝色圆圈显示了与O_pi_acc相互作用的配体氯原子的RF分布,α 定义为C-Cl键方向延长线的角度偏差,RF是 α (以10 °为 增量步长)的函数。在 0 °~10 ° 范围内(刚好是σ 孔方向)可以检测到最大值。RF值通常随着角度α的增加而减小,但在角度范围0−40°内,RF值明显大于1。在此范围内,RF值为1.45 ± 0.13。 我们从PDB推导的倾向性支持了Cl··O=C相互作用是有利的这一观点,并且我们发现,如果与之相互作用的羰基氧原子位于由C···Cl矢量的延长线上(Cl···O距离小于3.8 A)以及α=40°开角所定义的圆锥内,与蛋白结合位点中其它有利的分子间相互作用相比,Cl·O=C相互作用具有很强的竞争优势。
表4. O_pi_acc型蛋白羰基氧原子与不同配体卤原子相互作用的RF值和统计不确定性

aα = 0−40°角度约束的统计分析,其中α定义为C-卤素键延长线方向上的角度偏差(见图4)。方括号里的RF值为自举法得到的95%置信区间。
如果我们取相同角度范围(α=0−40°),探测其它卤素的相互作用统计数据,我们发现氟和重卤素之间具有明显的区别(表4)。 氟不能形成σ-孔穴,显示出统计学意义上的显著的RF小于1,而其它卤素RF大于1。尽管由于PDB中的较少实例导致的统计误差线对于溴和碘而言较大,但是很显然。RF与卤素尺寸大小具有明确的相关性。由于卤键在实验和理论上都得到了广泛的研究,令人欣慰的是,我们的统计方法与先前的研究结果一致,支持了除氟之外的卤素稳定σ-空穴相互作用的这一概念,这种相互作用随着原子尺寸的增加而增加。
表5. 就与芳香碳相连的配体氯原子而言,不重复的配体和不重复的靶标的有利相互作用类型、相关角度、RF值和LOS接触数目的统计

卤键案例研究结果表明,图3的概要图有助于初步了解优选的相互作用模式,但需要用相互作用角度进行更详细分析以提供更有意义的RF值。 因此,我们以10°的α增量步长对所有蛋白原子类型进行了角度扫描,从中提取出RF始终大于1的角度范围。对于配体氯原子而言,这产生了表5中总结的优选相互作用对象以及相关α范围。其中有几个问题值得进一步讨论。
极化的CH基团(C_ali_don)定义为与吸电子氧或氮原子共价键连接的CH基团,其角度依赖性如图4橙色圆圈所示。该曲线表明了最佳角度范围为40-90°,这完全不同于氯与O_pi_acc之间的相互作用行为。有趣的是,观察到的倾向性增加与卤素中σ-空穴的电子分布非常吻合:在C-Cl键的方向上带正电荷,而在其周围带负电荷[6]。我们的统计数据支持这一观点,具有静电互补性的蛋白官能团具有很高的倾向性,即富含电子的羰基氧原子在C-Cl键以及极化的CH基团方向上,它们的正电末端垂直对齐于羰基氧。如图4的两个示例所示,若α大于等于120°,则RF值通常会下降。这是因为在这个相互作用角度,蛋白-配体排斥相互作用越来越占主导地位。
原子类型C_ali_don可以解释为弱氢键供体。有趣的是,蛋白原子类型N_pi_don也发现了相似的有利角度范围α=40−80°,它代表了一类嵌入在π-体系中更强的氢键供体,比如酰胺骨架的NH基团(表2)。然而,仔细观察会发现,在本案例中,占优势地位的几何结构不具备氢键相互作用特征。根据氢键相互作用的几何结构特征,氯原子应该在π-体系的平面内,而不是在平面上方的一种相互作用(图S6)。
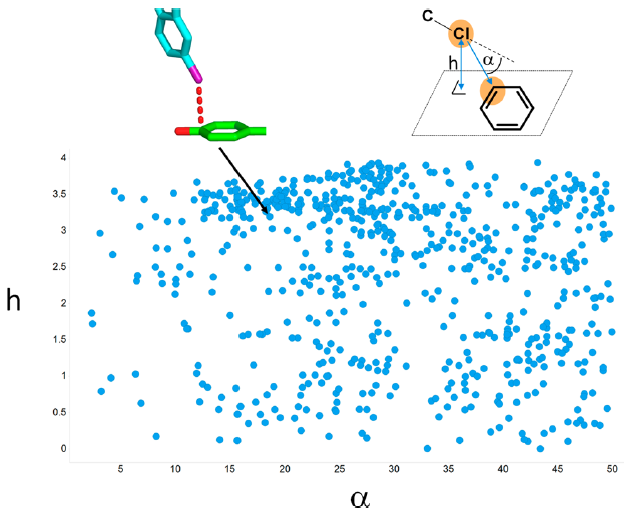
图5.与芳香碳连接的配体氯原子与蛋白原子类型C_pi_phenyl之间的LoS相互作用几何分布。高度h是氯原子到苯环平面的距离(单位Ångstrom),α是与C-Cl键延长线偏离角度。相互作用角度限制于0-50°,这是表5识别到的优势分布范围。大部分点聚集于高h值区,代表性例子有图示的FXa共晶结构PDB 3q3k41。
在丝氨酸蛋白酶Factor Xa的S1口袋中发现的一种相互作用被人们认为对某些几何排列具有很强的稳定化作用,其中涉及氯和溴原子与芳环的卤键相互作用[39,40]。实际上,如表5所示,我们发现一种连接到芳环的配体氯原子与蛋白原子类型C_pi_phenyl的相互作用(α= 0-50°),其具有统计学显着性的RF值(1.37 ± 0.12)。如果我们关注此范围内的α,并对氯原子到苯环平面的高度h绘图,我们会发现多种排列方式,但其中大多数会聚集于高h值处,其中C-Cl键会指向苯环(图5)。我们在158种不同的蛋白靶标的389种配体中发现了α在指定范围内的这种相互作用类型,这表明这种具有高度倾向性的相互作用并不限于FXa结合位点,而是发生在大量不同的蛋白环境中。
图6. 与芳香族碳相连的配体氯原子与蛋白原子类型C_pi_carbonyl间LoS相互作用的几何分布。α与β角的定义如图左上角所示。相互作用角度α限于0-90°,这与表5中识别到的优势范围是一致的。主要集中分布在β=α的直线周围。来自ephrin A2受体(左,PDB代码5ia3)44与Bace-1(右,PDB代码4djx)[43]的两个示例可以用来说明几何结构排列。
表5中氯原子的最高RF值(2.18±0.13)来自与蛋白原子类型C_pi_carbonyl的相互作用,后者是蛋白骨架酰胺或天冬酰胺/谷氨酰胺侧链羰基的碳原子。考虑到第二个相互作用角β,即C-Cl键矢量和蛋白羰基平面的法线之间的角度,揭示了两个角α和β之间的相关性(图6)。代表性示例说明了在分布稠密的区域中观察到的相互作用几何结构。这些几何结构可以多种多样:C-Cl键矢量可以几乎垂直指向由羰基及其相邻原子形成的平面(图6,左),C-Cl键矢量的取向还可以平行于羰基平面(图6,右)。我们发现,与正交取向(α=β= 0-30°时分布为11%)相比,平行排列的分布密度更高(30%以上的角度分布范围为α=β=60-90°)。这与先前对CSD[42]进行的数据库挖掘研究结果一致,这看起来像是Cl原子周围电子密度各向异性分布的另一种表现形式。一个有趣的例子是Bace-1与亚氨基乙内酰脲抑制剂的复合物结构,其中配体氯原子与蛋白S3口袋入口处的酰胺骨架碳原子发生了两次如此短距离的相互作用(图6,右)。配体中其它的氯原子导致抑制剂对Bace-1的Ki降低了5倍[43]。
另一个高倾向性相互作用,尽管例子相对较少,是氯取代基与精氨酸侧链中带正电荷的碳原子的相互作用(C_pi_pos,RF=1.59,误差线为1.01-2.22)。与图6示例BACE-1中看到的排布相似,其优选的相互作用几何结构是平行的。
表6. 键连于芳香碳原子的配体氟原子有利的相互作用类型、相关角度、RF值、LoS接触数统计数据、不重复的配体及其靶标。

由于氟取代基对结合亲和力、代谢,膜通透性与P-gp转运具有调节作用而在药物设计中大量使用[45,46]。我们将研究重点放在与芳环键连的配体氟原子上,PDB分析揭示了表6中列出的四个优势相互作用对象及其相关的几何结构(相关的汇总图,请参见图S7)。其中三种相互作用类型的贡献从α= 0左右开始,α= 0是C-F键局部偶极矩的方向。第一种相互作用对象是极化的CH基团(C_ali_don),其RF值为1.64±0.11,这可归因于有利的偶极空间取向,其中氟原子的部分负电荷指向极化的Hδ+—Cδ-键的部分正电荷。 在我们的统计数据中,我们将其视为首选的相互作用模式,但仅适用于与极化CH键的接触。对于由原子类型C_ali_apol为代表的弱极化CH基团,如此高的倾向性角度区域并不明显。与蛋白原子类型C_ali_don相互作用时,氟和氯之间的一个有趣差异是首选角度范围:氯的角度范围为40−90°(表5,αmax=75 °),氟的角度范围为0−70°(表6,αmax=5°)。这说明了两种元素有不同的电子特性。考察沿C−X键延伸方向 α=0−40° 的角度范围,我们会发现X=F的RF值为2.02 ± 0.23,X=Cl的RF值为0.97 ± 0.13,这表明氟原子可以特别强烈地稳定与C−F键偶极子方向上的极化C−H片段之间的相互作用。更重的卤素(溴和碘)的此种相互作用类型统计数据类似于氯的统计结果(见表S4和S5)。氟与氯统计数据之间的另一个区别是:在诸如O_pi_acc或C_pi_phenyl之类的富电子原子类型上,我们没有发现高倾向性的角度范围,考虑到氟的高电负性,这也并不奇怪。
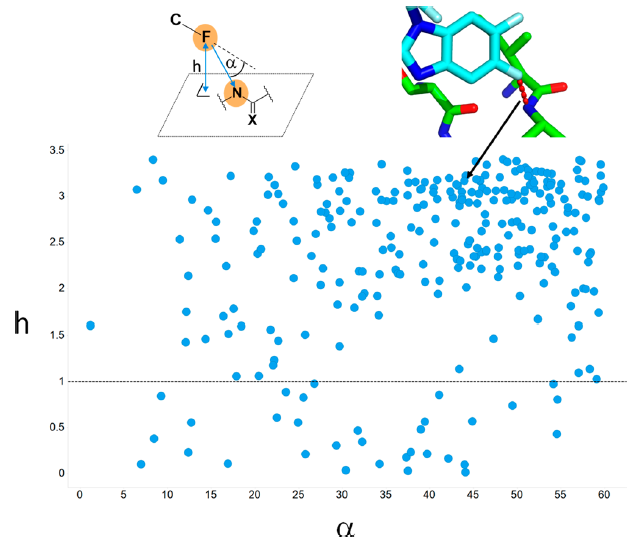
图7. 键连于芳香碳原子的配体氟原子与蛋白原子类型N_pi_don的LoS相互作用几何分布。高度h是氟原子到π-体系(X=C,O)N_pi_don平面的距离,α是与C-F键延长线的夹角。相互作用角α限于0−60°,为表6识别到的优势角度。h = 1.0Å的虚线将氢键相互作用特征(h小于1)与其它的空间排列区分开来。大部分点聚集于高的h值区,图中示例的相互作用几何结构为一个FXR复合物结构,其PDB代码为5q14[49]。
氟与蛋白质原子类型N_pi_don的有利相互作用区域为α=0-60°。这是由于氢键相互作用的高倾向性?还是相互作用的几何结构不同?在图7中使用的几何结构定义中,氢键几何结构中氟原子位于或靠近供体π-体系的平面,从而距离h值很小。可使用h=0-1Å的距离范围作为氢键的判据,发现仅11.3%的命中。与氯衍生物的分析结果(7.9%,见图S6)相比略高,但仍然相对较小。图7揭示了氟与强氢键供体N_pi_don的大多数相互作用具有倾向性的堆积几何结构,并且还不是氢键相互作用。尽管氟和强氢键供体之间的高水平量子力学(QM)计算显示出吸引的相互作用势[47],但我们并未发现弱氢键受体F与强氢键供体NH之间在蛋白结合位点内形成了具有竞争优势的氢键相互作用。相反,原子类型为N_pi_don的强NH氢键供体似乎更喜欢与较强的受体(例如O=C基团)发生氢键相互作用,而氟原子优先以倾斜方式与N_pi_don的π平面的弱极性侧面相互作用。与此相符,我们没有发现氟与其他氢键供体之间具有高度相互作用倾向性范围。我们的结论是,与更强的氢键受体相比,有机氟与N-H或O-H供体的氢键相互作用没有竞争优势,这与先前对小分子晶体结构的分析结果相符[48]。
对于氟而言,表6中的最高RF值是原子类型C_pi_carbonyl(RF = 2.37)和C_pi_pos(RF = 2.34),具有与氯原子相互作用相同的角度范围和αmax值(表5)。 Paulini等人[17]在以前的CSD搜索中也发现了C-F···C=O相互作用的高倾向性。
腈基的相互作用
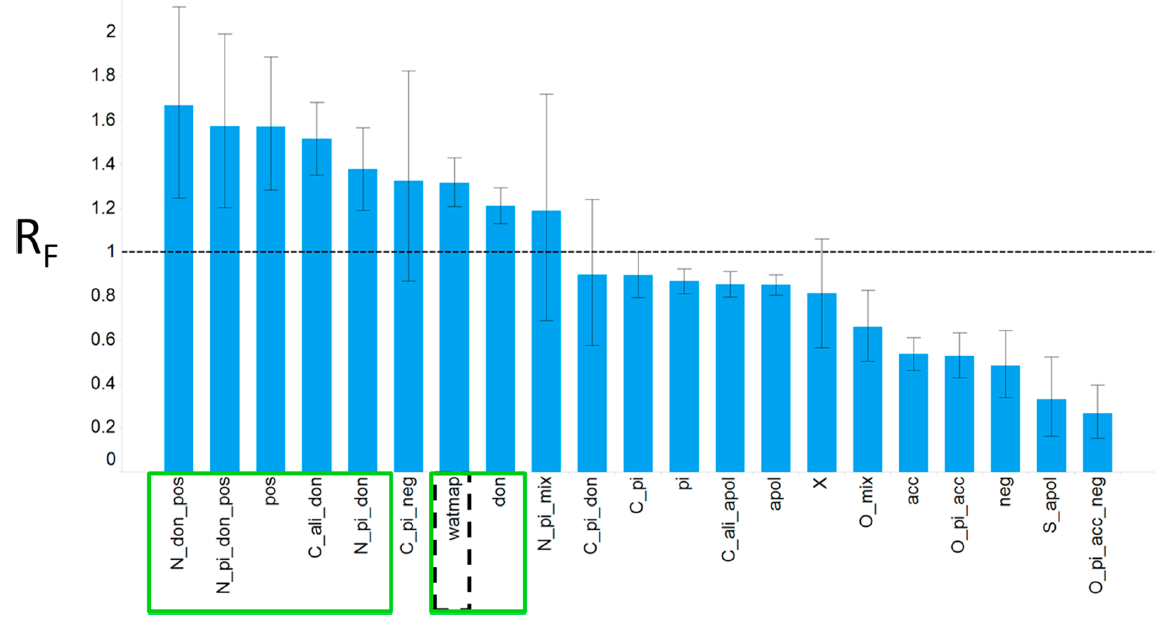
图8. 配体腈基中氮原子的RF相互作用统计数据图。若复合物中腈基碳原子与蛋白原子之间的距离较短(d 小于等于 2.25 Å),则意味着配体与蛋白发生共价键,此种情况应该予以排除。该注释的详细描述见图3。
腈官能团具有一个大的局部偶极矩,因此是一个令人感兴趣的小分子设计元素,安装后可与蛋白受体发生特定相互作用,同时也用于增加配体的极性[50]。使用我们的方法,可以回答两个问题:腈基在蛋白-配体复合物中的溶剂暴露到了何种程度以及它是否参与了特定的蛋白-配体相互作用?配体腈基有时被用作亲电试剂与半胱氨酸侧链共价相互作用[51,52]。由于这会导致不同相互作用模式掺杂在一起,因此我们将腈基碳原子和任何蛋白原子之间距离很短(d 小于 2.25 Å,这意味着共价键)的复合物结构从我们的分析中排除在外。对于与蛋白环境非共价相互作用的腈基,我们发现它倾向于与氢键供体以及正电荷相互作用,与负电荷、氢键受体以及非极性环境发生相互作用的频数比期望的随机发生要少(图8)。腈基也经常被发现至少部分暴露在溶剂中,292个蛋白−配体复合物就是这种情况,与原子类型“watmap”相互作用的RF值为1.32 ± 0.08。
表7.与配体腈基氮原子相互作用的有利类型、相关角度及RF值
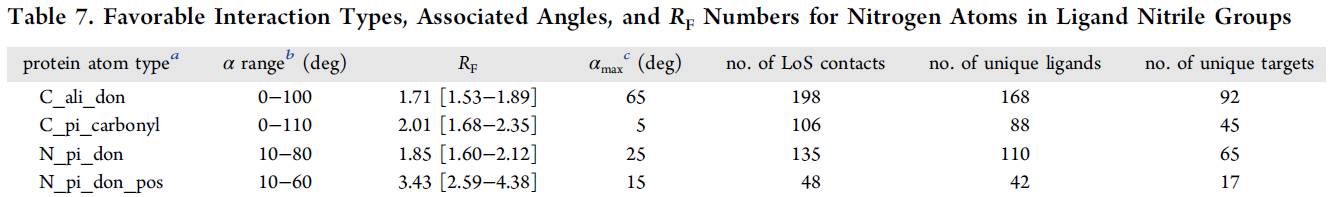
a蛋白原子类型的定义见表2。b相互作用角度α的可视化见图4, Cl用配体腈基的N替换了。角度范围确定方法描述见实验部分。c角度分析中以10°步长最大RF的中点(见图4)。

图9. 腈基氮原子与蛋白原子类型N_pi_don_pos之间的LoS相互作用几何分布。高度h是腈基氮原子到胍基π-体系平面的距离(单位为Å), α为CN三键延长方向上偏转角度。相互作用角度α限制于10−60°,这个角度是表7识别到的优势范围。 h = 1.0 Å的虚线将氢键几何结构特征(h小于1)与其它空间排列区分开。虚线上方的代表性相互作用几何结构有FXR复合物(PDB 1ld7)[54],虚线下方的代表性相互作用几何结构有AR复合物结构(PDB 3b5r)[55]。
考虑到腈基和蛋白质接触原子之间的相互作用角α,我们发现了表7中所示的原子类型和角度范围的高倾向性环境。与N_pi_don_pos(代表精氨酸残基中胍基尾部的氮原子)原子类型相互作用的RF值异常大,为3.43±0.95。虽然统计误差线相对较大,但这种类型的相互作用仍然存在于17个独特靶和42个独特配体组成的多样性数据集中。胍基可以通过电荷辅助氢键与腈基的氮氢键受体在平面内相互作用,也可以通过阳离子-偶极相互作用在平面上方相互作用。图9显示了这两种类型的排列都可以在PDB中找到,这有助于提高RF值。在图9的数据集中,可以发现几个例子,其中占优势的胍···腈相互作用显著地增强了对感兴趣靶标的亲和力。来自默克公司的一系列DPP-4抑制剂中,用更长的腈基取代了氟,使得IC50值改善了近100倍(图S8)[53]。氰取代基与DPP-4的主要相互作用是与Arg125的胍基尾部之间的短氢键(d=3.0 Å)。另一个腈基对提高结合力起到至关重要作用的例子来自一个法尼基转移酶项目,其中氰取代基与Arg702发生短的阳离子-偶极相互作用(图S9)[54]。有趣的是,原论文的作者评论说,“最接近氰基的氨基酸侧链是β亚基中的Arg702。它的胍基与腈基氮相距约3Å,但这两个基团的相对几何结构似乎与直接相互作用不一致”。我们的统计分析表明,在氢键和阳离子-偶极几何结构中,胍···腈的相互作用确实应该被认为是潜在的非常有利的直接相互作用。在我们看来,这种专一靶向精氨酸侧链(已经理想地通过其他相互作用进行约束以避免额外的熵惩罚)是一个被忽视的提高靶标结合亲和力的优化策略。
嵌入在π-体系内的强氮氢键供体(N_pi_don)在与腈基氮发生相互作用时也被发现具有高RF值(RF=1.85 ± 0.27)。与胍盐的情况类似,这种相互作用对象在平面内也具有氢键势,在平面上方具有较低的极性相互作用表面,根据图S10,这两种类型的相互作用几何结构都会发生。
磺酰基的相互作用

图10. 砜(上)与磺酰胺(下)子结构中氧原子RF相互作用统计图,注释参见图3。
实验测得的氢键碱度将磺酰基列为中等强度的氢键受体[56]。基于CSD和PDB搜索,提出了氢键受体和疏水基团的双重特性假设[42],在现有的蛋白−配体复合物中磺酰基仅位于纯非极性环境中[57]。我们对砜和磺酰胺中氧原子的优选环境的研究结果显示,它与经典氢键供体相互作用的倾向性很高(图10)。更强受体的砜(N_pi_don,1.53;watmap,1.45)比磺酰胺(N_pi_don,1.54;watmap,1.18)的RF值稍高一些。与CH基团的相互作用,只有在它们极化时(C_ali_don,磺酰胺和砜的RF分别为1.66和1.23)才会发生,此时RF值大于1;而对非极性碳原子(C_ali_apol,磺酰胺和砜的RF分别为0.69和0.60)明显没有竞争优势。配体磺酰基片段与极化的CH基团发生三次相互作用的一个例子来自组织蛋白酶S(图S11)。还应提到的是,与原子类型O_mix(其为丝氨酸或苏氨酸侧链中的羟基)相互作用的RF值明显低于1,尽管它们具有氢键供体官能团。我们相信这是我们的隐式氢方法的结果,在该方法的结合位点里仅O_mix的氢键受体官能团暴露用于配体结合,从而降低与受体磺酰基氧原子接触的RF值,而OH供体与蛋白发生分子内相互作用。
表8. 与配体磺酰基氧发生有利相互作用的类型、角度以及RF值a

a磺酰基(Sulfonyl)代表砜、磺酰胺、磺酸酯/盐的一部分基团SO2。b蛋白原子类型的定义见表2。c相互作用角度 α的可视化见图4,其中C-Cl用SO2代替。角度范围如何确定请参见实验部分。d角度分析中以10°步长最大RF的中点(见图4)。
表8列出了一般情况下磺酰基(即作为砜、磺酰胺或磺酸盐的一部分)的优势相互作用类型与角度范围。我们发现了不同类型的氢键供体,若这些供体嵌入在π体系中,则是优势的氢键几何结构。这与在氟上观察到的优势相互作用几何结构有着质的不同(图7)。值得注意的是,与带正电荷的蛋白原子(N_don_pos,N_pi_don_don_pos)相互作用也会有高RF值,这些蛋白原子分别是赖氨酸和精氨酸残基的氨基和胍基尾部。有趣的是,这种相互作用不仅发生在带负电荷的磺酸盐中。中性的磺酰胺与蛋白N_don_pos相互作用的RF=2.21±0.62(α=0−70 °),这表明该配体片段可能是与结合位点中赖氨酸侧链的有益相互作用对象。存在这种相互作用的例子来自不同的结合位点,比如BRD4(PDB 5uer)、PPARγ(PDB 5tto)[58]、或 β-内酰胺酶(PDB 3o88)[59]。
芳香硫的相互作用
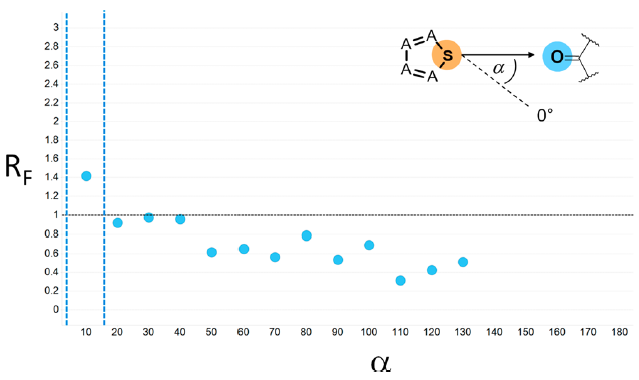
图11. 与蛋白原子类型O_pi_acc相互作用的五元芳香环中硫原子的相互作用角度RF统计数据。x-轴为10°的角度增量步长的角度α,定义为邻居原子到S原子之间键延长线(虚线)的夹角。因为硫原子有两个邻居原子,我们取角度小的那个。RF=1的水平线意味着该线上RF值为随机期望值。虚线竖直线定义了有利的角度范围。
σ-空穴键(σ-hole bonding)的一种变体是用硫原子而不是卤素原子作为σ-空穴供体,它与分子间识别以及基于结构设计的相关性备受争议[10,12]。先前对CSD数据库的研究表明,硫原子和羰基氧原子之间的相互作用可以具有很强的分子内构象导向效应[60]。此外, 从蛋白-配体复合物中收集了大量的例子,其中观察到的几何结构表明:在二价硫原子的σ-空穴(σ-hole)与给电子的Lewis碱之间存在有利的相互作用[11]。由于σ-空穴预计位于硫σ键的延伸方向上,角RF统计数据可以揭示这种类型的相互作用(图11)。与氯原子的类似曲线图(图4)相比,有两个明显的区别:首先,与氯的RF=2.4相比,硫的最大RF=1.4,信号明显较弱;其次,在零假设线(RF=1)以上的RF值,硫比氯的角度分布范围更小,分别为α=0−10° vs α=0−40°。总的来说,我们的统计数据表明,发生在蛋白-配体复合物中的分子间硫 σ-空穴相互作用频次仅比期望的偶然发生频次高那么一点点,而且这仅适用于非常窄的角度范围内。我们的发现质疑了硫原子与蛋白羰基的σ-空穴键是蛋白−配体结合位点中一种特别重要的相互作用假设。一个影响我们观察结果的因素是,许多含有芳香硫片段的PDB配体能够形成分子内S···O/N硫族元素键(chalcogen bond)[60],这将显著地降低分子间相互作用倾向。理论上,σ-空穴应该随着芳香环中极化氮原子的增加而增加,CSD搜索显示噻唑的分子间S··O或S··N相互作用的命中率(9.3%)比噻吩(4.7%)高大约2倍[61]。相比之下,我们的统计数据并没有显示这两个子结构的RF值有明显差异;但是,噻唑基团在PDB中的覆盖率相对较小。更多的含有噻唑配体的蛋白结构将极大地有助于以更高的统计确定性分析这种差异。
表9. 与五员芳环硫原子有利的相互作用类型、角度以及RF值

a蛋白原子类型的定义见表2。b相互作用角度 α的可视化见图11,角度范围如何确定请参见实验部分。c角度分析中以10°步长最大RF的中点(见图11)。
芳香硫喜欢优先与哪些蛋白质环境相互作用呢?从优势的相互作用类型与角度列表(表9)以及角度忽略的概要图(图S12)中可以看出,大于1的RF值仅适用于与碳原子的密切接触,这些碳原子要么是不同π体系的一部分,要么含有极化氢原子。图12说明了一些优势的几何结构,其中包括与芳环和酰胺基团的堆积相互作用。
结果与讨论
我们提出了一种新的方法来挖掘蛋白-配体复合物晶体结构数据,使用两个重要的成分:(a)视线过滤器以去除不需要的分子间接触;(b)基于结合位点暴露表面积可靠的统计模型。由此,对于在基于结构的药物设计中感兴趣的一组配体基团,我们能够推导出以RF值表示的蛋白原子类型的相互作用倾向。 这些相互作用倾向可以作为接触基团之间的相互作用角度的函数来计算,这使得我们能够检测优选的相互作用几何结构。大于1的高RF值意味着特定相互作用类型比偶然预期的更经常发生。这表明,在所确定的几何结构范围内,这种相互作用不仅具有吸引力,而且相对于在结合位点中形成的所有可能的其它有利的相互作用来说都具有高度竞争优势。图12给出了本报告中研究的配体官能团的所有具有高RF值的相互作用的代表性实例。我们相信这里总结的相互作用和相关的角度范围对于结构指导的亲和力优化特别有吸引力。

图12. 与氯原子(表5)、氟原子(表6)、腈基氮原子(表7)、磺酰基氧原子(表8)以及芳环硫原子(表9)高RF值的蛋白-配体相互作用概述。对于每种相互作用类型,给出角度分析中(见图4)RF值最高的代表性例子。蛋白原子类型的定义见表2。RF值指的是指定范围。相互作用角度α由配体接触原子、以及其共价连接的邻居原子和蛋白接触原子共同定义。
我们将这种方法应用于研究那些被人认为非经典的、据推测是有利的蛋白-配体相互作用。其中某些相互作用,例如氯以及重卤素与羰基氧原子的σ-空穴键(σ-hole bonding),我们为有利的、具有竞争优势性的相互作用找到了可靠的支持,许多不同的结合位点均存在这样的相互作用。对于其它类型相互作用,例如以氟作为受体、NH作为氢键供体的氢键,我们的研究没有证据表明这种氢键相互作用发生的频率高于期望偶然发生。有趣的是,高水平的QM计算预测该相互作用具有吸引的相互作用能势。我们怀疑产生这种差异的部分原因是由于在结合位点外的孤立模型系统中执行了QM计算,从而给出了在气相或溶剂模型中的相互作用强度信息,但不给出在蛋白结合位点中的竞争性的信息。值得注意的是,我们发现了具有非常高RF值的其它相互作用,例如腈基与精氨酸侧链的胍基尾部之间或磺酰胺氧原子与赖氨酸残基的氨基之间存在相互作用,据我们所知,这些相互作用还没有被广泛地用于结构指导设计。
显然,大量其它令人感兴趣的相互作用模式可以用这种方法进行研究,这将是未来工作的主题。此外,有必要对低RF值的相互作用类型进行深入研究,识别不利的或非竞争性的蛋白−配体接触是值得的。在药物发现项目的早期从共晶体结构或模拟的结合模式中识别不利的相互作用可以显著地减少为实现与所需靶标结合而进行优化的循环次数。
实验部分
蛋白-配体结合位点
不带氢原子的蛋白结合位点以Mol2格式从Prosis62提取得到,Prosis是注释过的PDB,并存为SQLite数据库。除了金属、常见的离子之外,所有的HET分子都生成为数据库条目(2017年11月30日)。这些HET基团至少含有5个原子,最多含有100个原子。在HET 6Å半径范围内所有的分子都包含在各自的数据库条目中。该数据库包括了从74436个PDB条目导出的276065个结合位点。
同一蛋白-配体组合的晶体结构有时被几个实验室使用稍有不同的蛋白结构或结晶条件来解释。例如,在PDB中存在51个牛胰蛋白酶与苯甲脒的复合物结构。为了避免对“相同”的蛋白-配体复合物的相互作用进行多次计数,我们应用了冗余过滤器。每个数据库条目同时用结合位点中蛋白的UniProt[63]标识符以及结合位点中HET的canonical SMILES来注释。具有相同UniProt-SMILES组合的数据库条目,仅保留具有最佳分辨率的条目。除了冗余过滤器外,我们排除了分辨率低于2.5Å的结构以及结合位点里有多个能与配体SMARTS查询相匹配的HET基团的结构。后一种过滤器只应用于少数结合位点,但有助于我们工作流中的统计分析。在下文中,结合位点中的中心HET基团被称为配体。我们注意到,对于更一般的相互作用搜索,除了在本研究中进行的那些,具有辅酶(cofactor)的晶体结构可能会出现偏差。这是因为辅酶结合位点环境在许多不同的蛋白质中通常是很保守的。因此,对于与辅酶部分匹配的配体官能团的查询,需要更精细的冗余过滤器。
视线相互作用(Line-of-Sight Interactions)
与泰勒的方法类似,我们在分析中只考虑了视线(line-of-sight,LoS)相互作用。LoS概念的实现如图1所示。匹配配体SMARTS查询的配体基团原子L与蛋白质原子P之间的所有分子间LOS相互作用距离为d,

其中rL与rP是取自Bondi[38]的vdW半径,x是距离增量。因为我们想要研究许多种不同的相互作用类型,其中一些具有超过范德华半径之和的稳定贡献,因此我们将距离截断增加了x=0.5Å。这还有一个额外的好处,即与x=0Å相比,统计误差线要小得多,这是因为更多的接触被考虑进去了。
我们知道van der Waals半径随隐式氢原子数[64]以及原子上形式电荷的程度而变化[65]。Bondi半径使用了一个近似值;然而,我们也没有简单的解决办法。例如,选择增大的半径将引入新的模糊性,特别是对于氮和氧原子,这是由于不可避免的氢键以及PDB结构中配体的潜在不准确的原子和键类型判断。我们还注意到,在之前的一项关于CSD数据的RF相互作用统计的研究中,比较了三组不同的半径,包括Bondi值,得出了相同的结论[36]。这使我们相信,使用Bondi半径是这本研究的适当近似值。
相互作用频数比(Ratio of Interaction Frequencies)
类似于Taylor的方法,对于给定的一对配体原子类型TA和蛋白质原子类型TB,将这种相互作用发生的被观测到的频数除以随机期望的频数来推导出相互作用倾向,即所谓的频数比RF(TA,TB):

出现频数观测值ΣO(TA,TB)i是通过计算结合位点数据库中原子类型TA和TB之间的所有LoS接触总和而获得,其中TA与TB需满足方程(1)扩展范德华截断的要求。随机期望频数是方程(2)的分母,是蛋白原子类型B的暴露表面积S(TB)与配体接触的总暴露表面积S(Total)比值然后在包含原子类型TA和TB的所有结合位点i上求和。对于这些表面的计算,考虑了结合位点中除配体以外的所有原子,但是仅对满足方程(1)的距离标准的那些原子进行求和。我们用溶剂可及表面来近似暴露表面,溶剂半径为1.4Å。 最后,方程(2)中的数目N是由TA型配体原子在结合位点i形成并满足标准1的LoS接触的数目。RF(TA,TB)值为1.0意味着TA与TB之间的相互作用以与偶然期望的频数相同的频数被观察到。
RF(TA,TB)的不确定性是通过使用10万个统计估计值自举所有包含TA和TB原子类型的结合位点的集合来估计的。将所得分布的第2.5百分位和第97.5百分位的RF值定义为95%的置信区间的下限和上限。
许多相互作用都是强方向性的,例如σ-空穴键,我们认为在额外的角度约束下进行分析是值得的。因此,我们不仅考虑LoS接触的距离,还考虑由配体接触原子、其共价相邻原子和蛋白接触原子定义的相互作用角α(有关α的定义,请参见图4)。在方程(2)中,ΣO(TA,TB)i为既满足方程(1)的距离约束又满足角度约束的相互作用的观察出现频数,而N为由同时满足距离约束与角度约束的结合位点i中的TA型配体原子形成的LoS相互作用的总数。
通过首先计算角度增量αi为10°的RF值并标记增量,确定每种接触类型的相互作用角α的优选范围,以满足以下标准:
- RF(αi)大于等于1.0
- RF(αi)大于等于0.95 if RF(αi±1)大于等于1.0
- 连续角增量数大于1
采用额外的统计显著性检验以确保所提取的角度范围的RF在95%置信区间内大于等于1。 此外,我们仅报道了当基于结合位点总体上的暴露表面积的预期命中数大于10时的优选相互作用范围(等式2中的分母)。
水的处理
PDB结构中水分子的位置通常是不确定的,因此,在我们进行分析之前,所有的水分子都被删除。取而代之的是使用水映射表(表3),该表绘将预期平均的显式水接触数映射到结合位点中计算的配体原子溶剂可及表面积(SASA)。本表由四个不同的具有极性配体原子的高分辨率(R小于等于1.5 Å)数据集(表S13)使用以下流程得到:(1)对于每个数据库条目匹配分辨率和配体子结构约束,提取与显性水分子接触的LOS数量和去除显性水分子后的SASA;(2)对于每个水接触数量(0、1、2、3)的SASA值分布,计算中值SASA; (3)最后的SASA范围通过取中值的中间点并对所有四个配体子结构求平均值来确定。表S13和图S14分别显示了极性配体原子的四种子结构定义和观察到的水统计数据说明。
在包括水分析的情况下,等式2的分母中的表面积包含表示暴露于水的附加项,其由结合位点中配体的SASA来近似处理。应注意的是,由于SASA测定中缺少角度信息,在本研究中有时无法将特定角度分析法用于水接触的分析。


