
Flare™ V12 发布:FEP 计算与基于结构的方法显著改进
摘要:Flare V12 正式发布,专注于提升计算药物发现中自由能微扰(FEP)方法的效率、准确性与适用范围。新版本引入断环变换支持,可一步完成骨架跃迁、环尺寸调整等复杂分子修饰。新 FEP 引擎 SOMD2 搭配"双系统"链接,使计算速度提升超过 2 倍(如 BACE 蛋白单次变换从 8 小时降至 3.8 小时)。集成 Hamiltonian 副本交换与 REST2,改善构象采样与收敛性;结合 GCMC 水采样技术,更精准描述溶剂效应。此外,Flare V12 扩展了分子动力学模拟范围,支持共价配体、非标准残基及翻译后修饰的自动参数化。分离式计算功能增强,可随时取消并获取中间结果,提高长时任务灵活性。这些更新帮助研究人员更高效地开展复杂药物设计。
Flare™ V12 发布:FEP 计算与基于结构的方法显著改进
我们很高兴地宣布 Flare V12 发布,该版本引入了一系列新功能和增强功能,旨在提高计算药物发现工作流程的效率、准确性和覆盖范围。
本次发布专注于推进自由能微扰(FEP)方法学,同时扩展了可靠建模的生物体系范围。这些更新使研究人员能够探索更复杂的设计挑战,更快地生成结果,并在整个药物发现管线中做出更明智的决策。
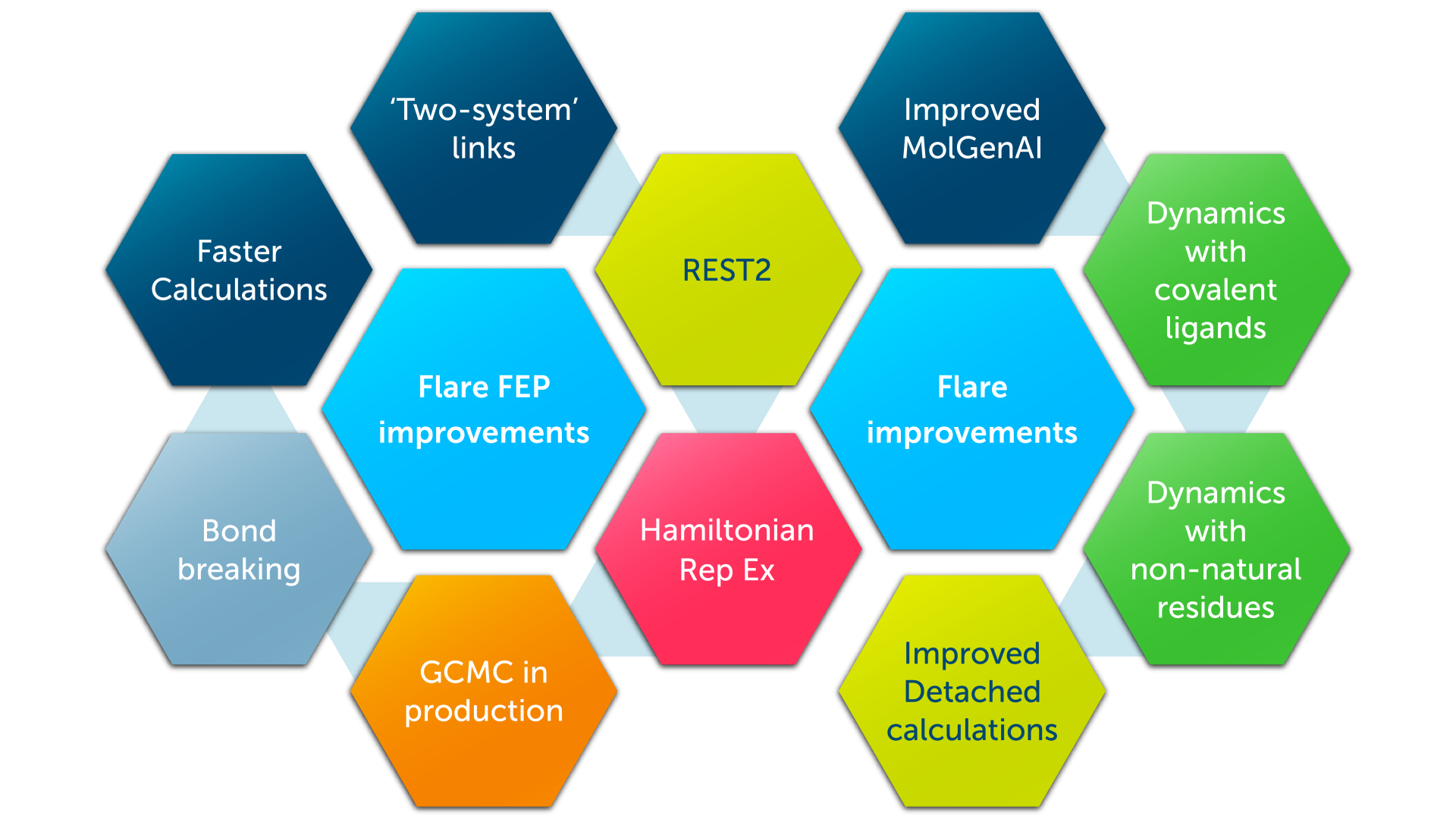
图 1. Flare V12 在 Flare 的基于结构和基于配体的方法中引入了重大改进
显著增强的相对 Flare FEP
本版本引入了相对 Flare FEP 适用性领域的重大扩展,包括断环(关环/开环)变换,以及更快的计算速度和更高的预测准确性。
在单次转化中模拟复杂的分子变化
Flare V12 引入了对断环变换(ring-breaking transformations)的支持,实现了骨架跃迁、环尺寸修改、饱和环与不饱和环之间的相互转换,以及环状结构与开链基团之间的转换。换句话说,它使得在单条变换路径中模拟复杂的分子变化成为可能。
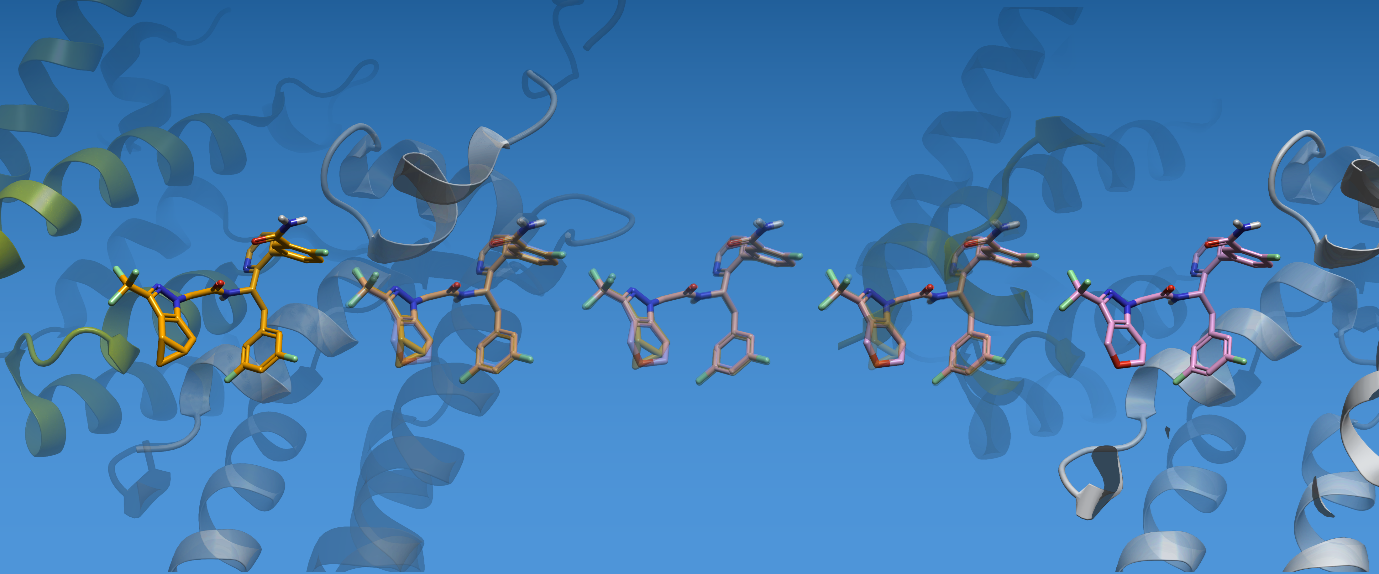
图 2. Flare FEP 中的环修饰现在更易实现,且可一步完成
Flare FEP 利用 Morse 势能代替普通键势能,让正在发生炼金术变换的化学键可以”断裂”,从而使相关原子能够拉开到很大距离而不会在 lambda 计算中出现奇点(势能发散),进而实现了环的修饰(如开环、关环等复杂变换)。环变换的一个示例如下图所示:
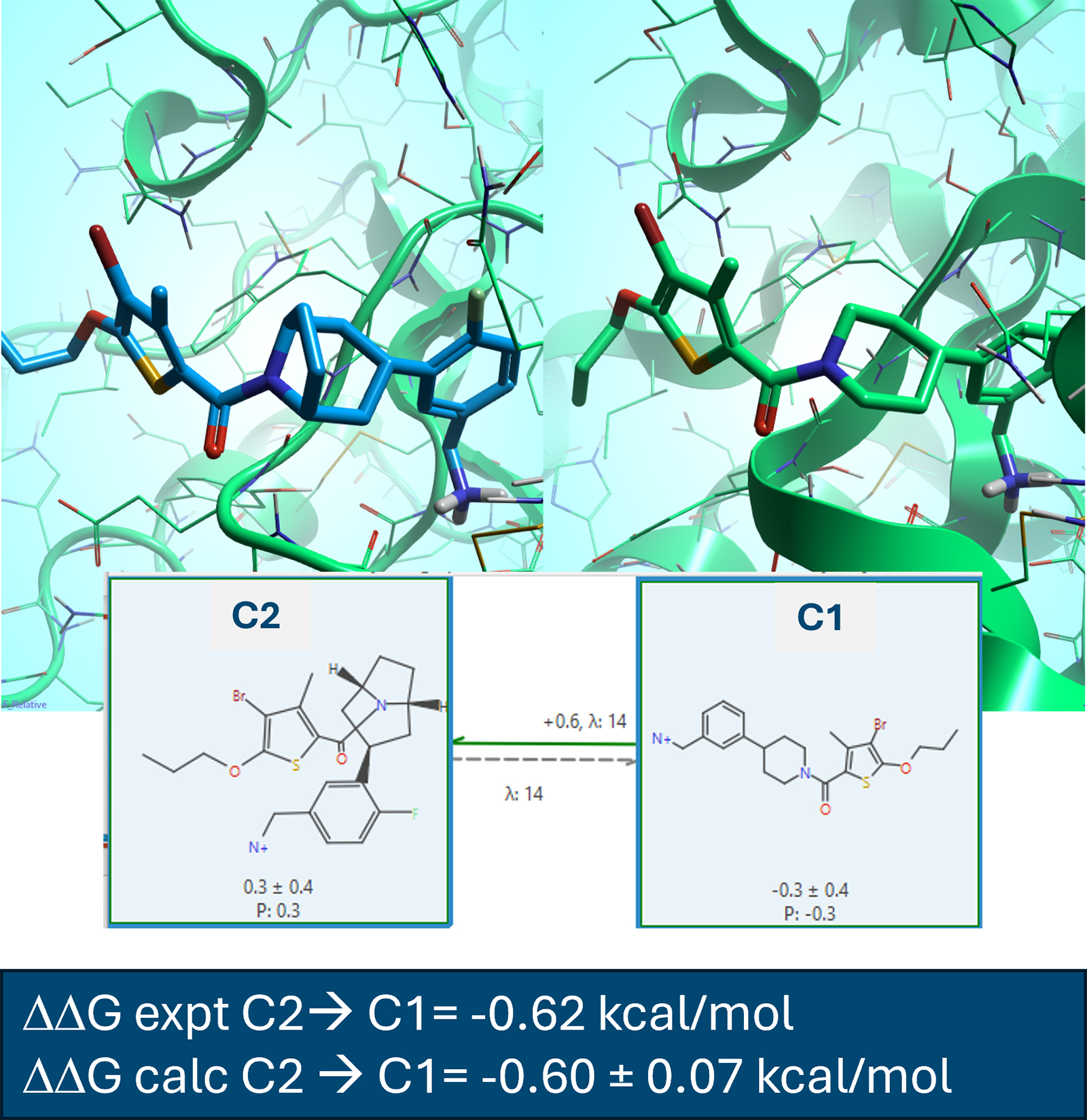
图 3. 来自 [1] 中 的TPSB2 蛋白质数据集示例。使用 SOMD2 计算得到的 C2 → C1 变换的 ΔG 为 -0.60 ±0.07 kcal/mol,与实验 ΔΔG = -0.62 kcal/mol 吻合良好。
借助新 FEP 引擎和”双系统”链接实现更快速的计算
Flare V12 配备了一个用于相对 FEP 的新 FEP 引擎(SOMD2),性能显著提升,计算速度提高约 20%。新引擎与新的”双系统”FEP 链接相结合,可实现超过 2 倍的计算速度提升。
通过”双系统”FEP 链接,模拟从变换的两个终态同时开始,并向相邻的 λ 窗口推进,在炼金术路径的中间汇合。为确保该路径上的充分采样和收敛,采用了副本交换技术,允许窗口之间交换构型,从而改善状态之间的重叠。
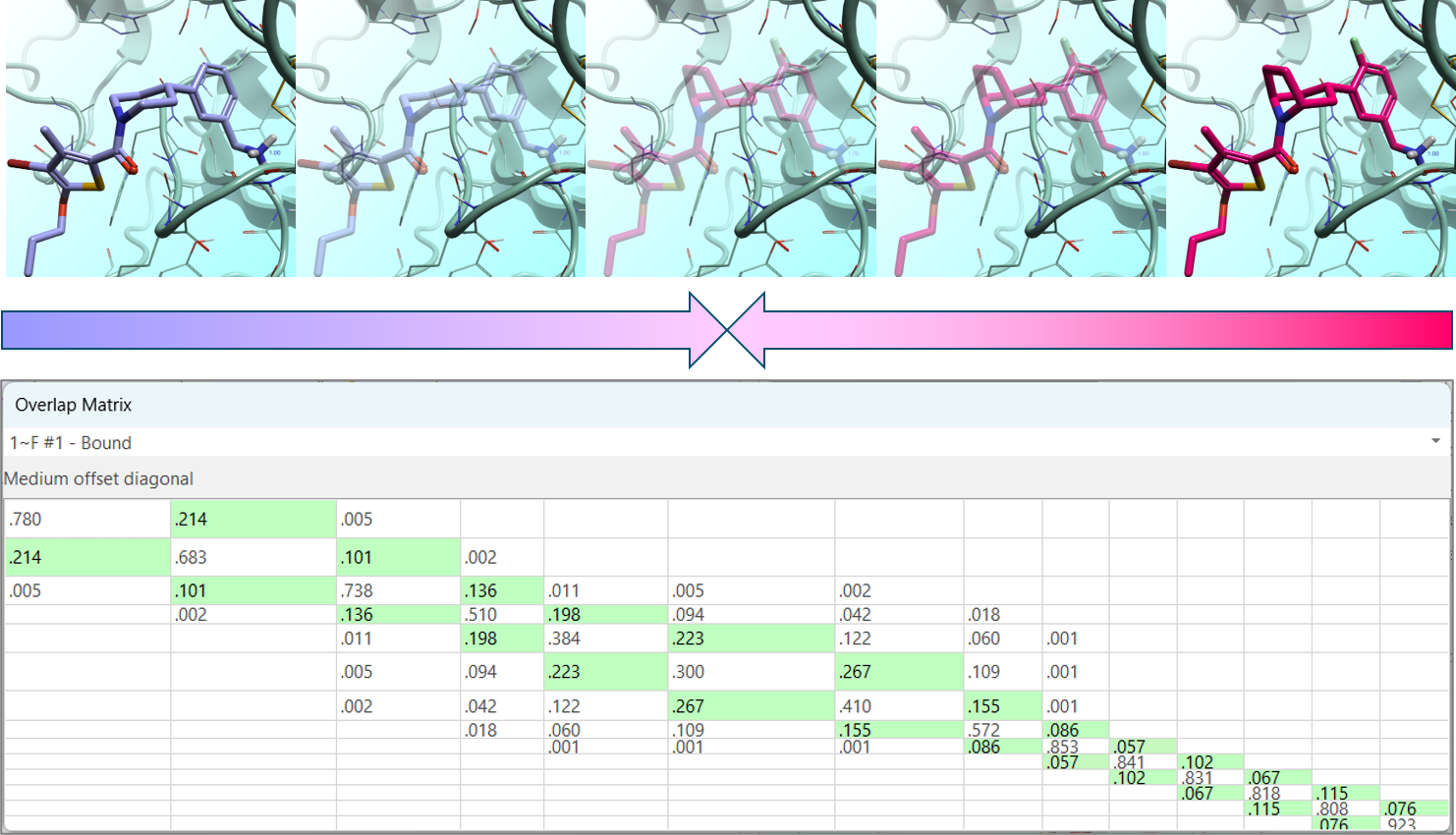
图 4. 通过”双系统”FEP 链接,模拟从变换的两个终态同时开始,并向相邻的 λ 窗口推进,在炼金术路径的中间汇合。副本交换用于确保充分的采样和收敛。
通过将先前 FEP 引擎所需的独立正向和反向扰动(molA → molB 和 molB → molA)合并为单一过程,Flare V12 提高了统计效率,同时将完成一次变换所需的时间减半。
例如,在入门级 GPU(AWS g4dn.xlarge,NVIDIA T4;约 390 个残基,默认计算选项)上,BACE 蛋白质中的单次 FEP 变换在 Flare V11 中大约需要 8 小时完成。在可比条件下,Flare V12 中相同的变换大约在 3.8 小时内完成。
启用 GCMC 水采样可以在溶剂效应至关重要的体系中显著提高准确性,额外计算成本通常约为 20-25%。
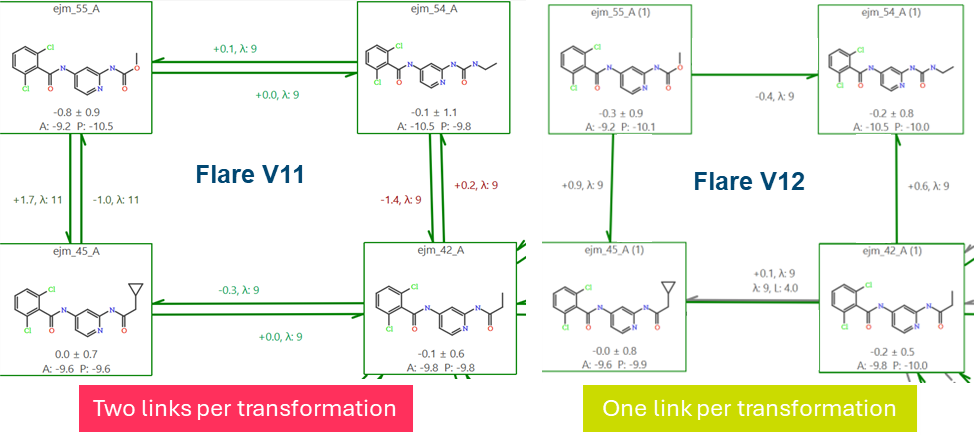
图 5. 通过双系统链接和新的 SOMD2 引擎(Flare V12 中默认启用),计算时间减半。
借助副本交换和 GCMC 提高预测准确性
将副本交换集成到 FEP 工作流程中,可以使研究体系探索更广泛的构型空间,改善收敛性,从而获得更可靠的自由能估算,特别是对于柔性或具有挑战性的体系。Flare V12 现已同时支持 Hamiltonian 副本交换 2 和溶质升温副本交换(REST2)3。
Hamiltonian 副本交换通过评估体系的势能并采用 Metropolis 接受准则,尝试在相邻的 λ 窗口之间”交换”构型。因此,在一个 λ 窗口中采样的构象可以传播到其他窗口,改善相邻状态之间的重叠,从而提高自由能估计的整体效率。
REST2 引入了一个新的维度——有效温度。通过提高分子中炼金术区域的温度,可以采样到更多的构象,使体系能够采用原本无法探索到的构象。
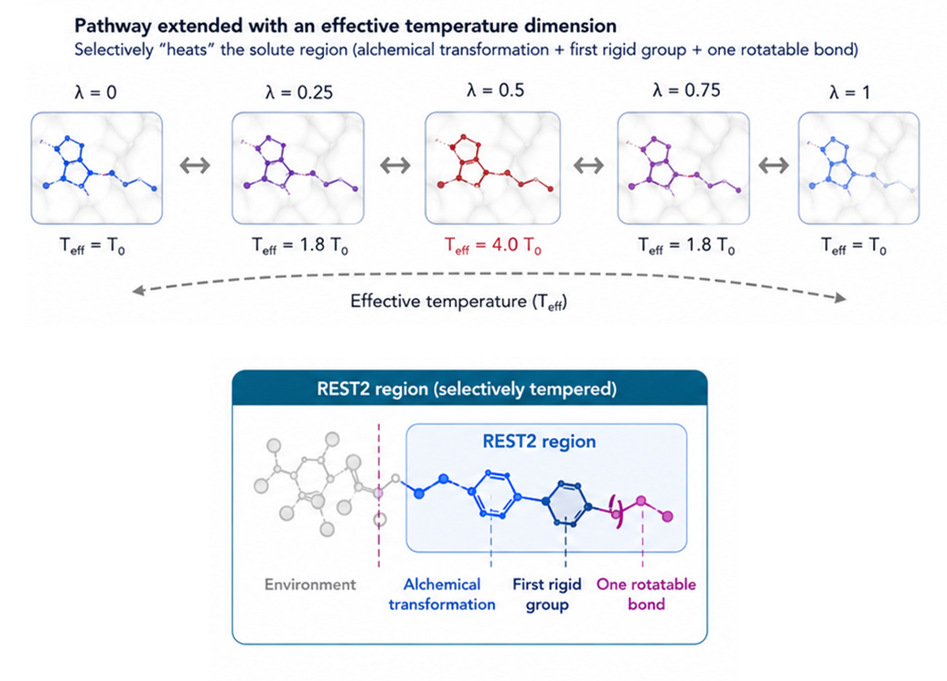
图 6. REST2 工作原理示意图。分子的一部分被加热,使得 Flare FEP 能够采样到室温下无法观察到的构型。
在 FEP 计算中启用 GCMC 水采样 4 可以在溶剂效应至关重要的体系中显著提高准确性,因为它能够更准确地描述溶剂行为。这种方法对于具有不易被本体水进入的深埋结合位点的体系尤为重要。GCMC 利用 Metropolis 准则,尝试在 GCMC 球体内插入水分子,从而更真实地描述炼金术区域周围的溶剂环境。此外,在水分置换有助于结合的情况下,使用 GCMC 的预测也更加准确。
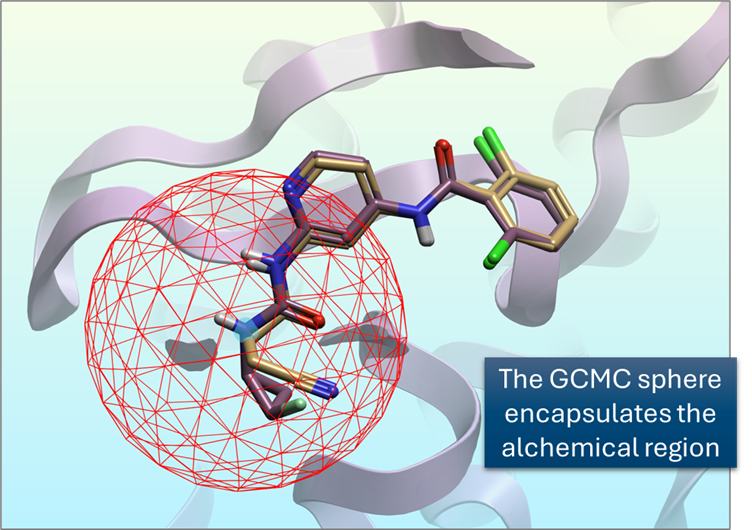
图 7. 生产运行中的 GCMC 确保结合位点的溶剂环境真实可靠,特别是在使用标准分子动力学难以采样水占据率的区域。
共价配体和非天然残基的分子动力学模拟
Flare V12 中分子动力学的适用性领域得到了显著扩展,支持共价配体以及肽配体和靶蛋白中非标准残基(包括翻译后修饰)的模拟。
Flare V12 引入了对修饰残基参数化的支持,使得在标准工作流程中能够处理包含翻译后修饰(PTM)、非标准氨基酸和共价配体的蛋白质。这在许多情况下消除了手动开发参数的需要,并能够无缝地集成到分子力学模拟中。
这得益于基于 Open Force Field 倡议中开发的翻译后修饰处理方法 5 的自动化工作流程。在该方法中,蛋白质和小分子力场组件被结合使用,以确保修饰后的残基与其周围的蛋白质环境完全兼容。与标准氨基酸共享的主链项源自 AMBER FF14SB 力场,而其余参数则使用 OpenFF Sage 力场分配。原子电荷使用 AshGC 电荷模型生成,为后修饰体系提供一致的描述。
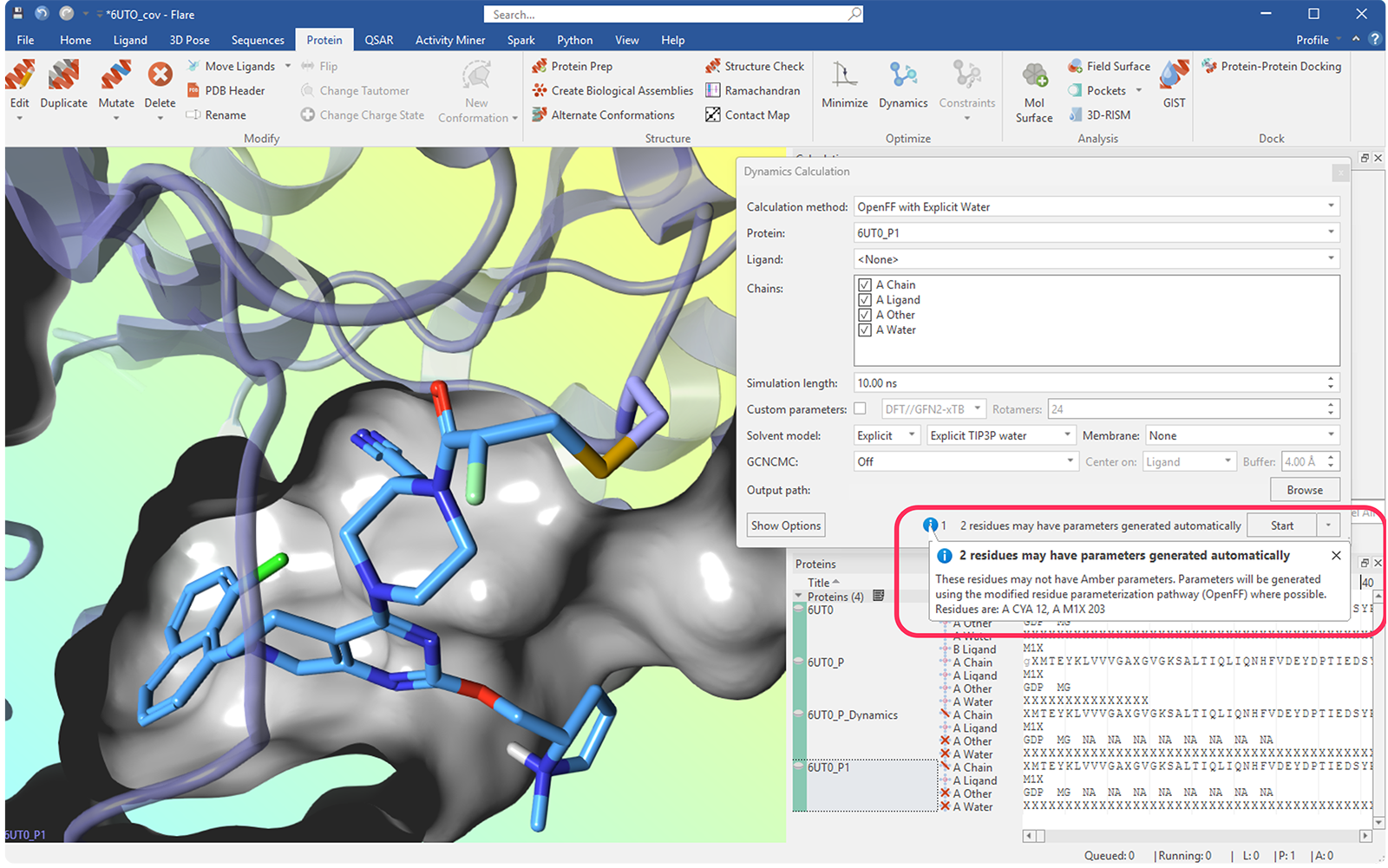
图 8. 在动力学计算面板中识别出一个与 CYS 12 结合的共价残基。Flare 通知用户它将自动进行参数化。
除了这些建模改进外,还对动力学工作流程进行了优化,使其更加流畅。当使用编辑功能修改残基时,其名称现在会在退出编辑器时自动更新。这避免了修饰后的残基保留标准氨基酸名称而导致动力学计算错误的问题。因此,体系准备更加稳健,所需的手动干预也更少。
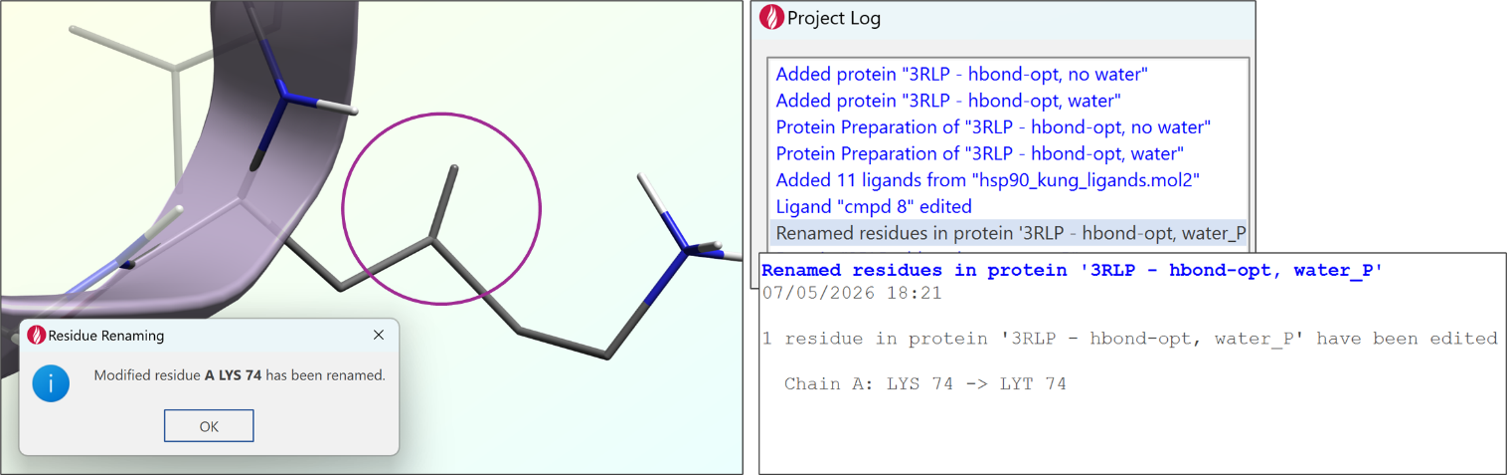
图 9. 修饰后的 LYS 被重命名为”LYT”,以避免分子力学模拟中出现问题。名称更改会在项目日志中报告。
增强的分离式计算用户体验
Flare 的最新版本对分离式计算的处理进行了多项改进,为长时间运行的作业提供了更大的灵活性和控制力。现在可以随时取消计算,并且截至取消时生成的任何结果都可以直接在 Flare 中下载和分析。这使得可以更有效地监控进度,并在无需等待计算完全完成的情况下做出更早的决策。
—
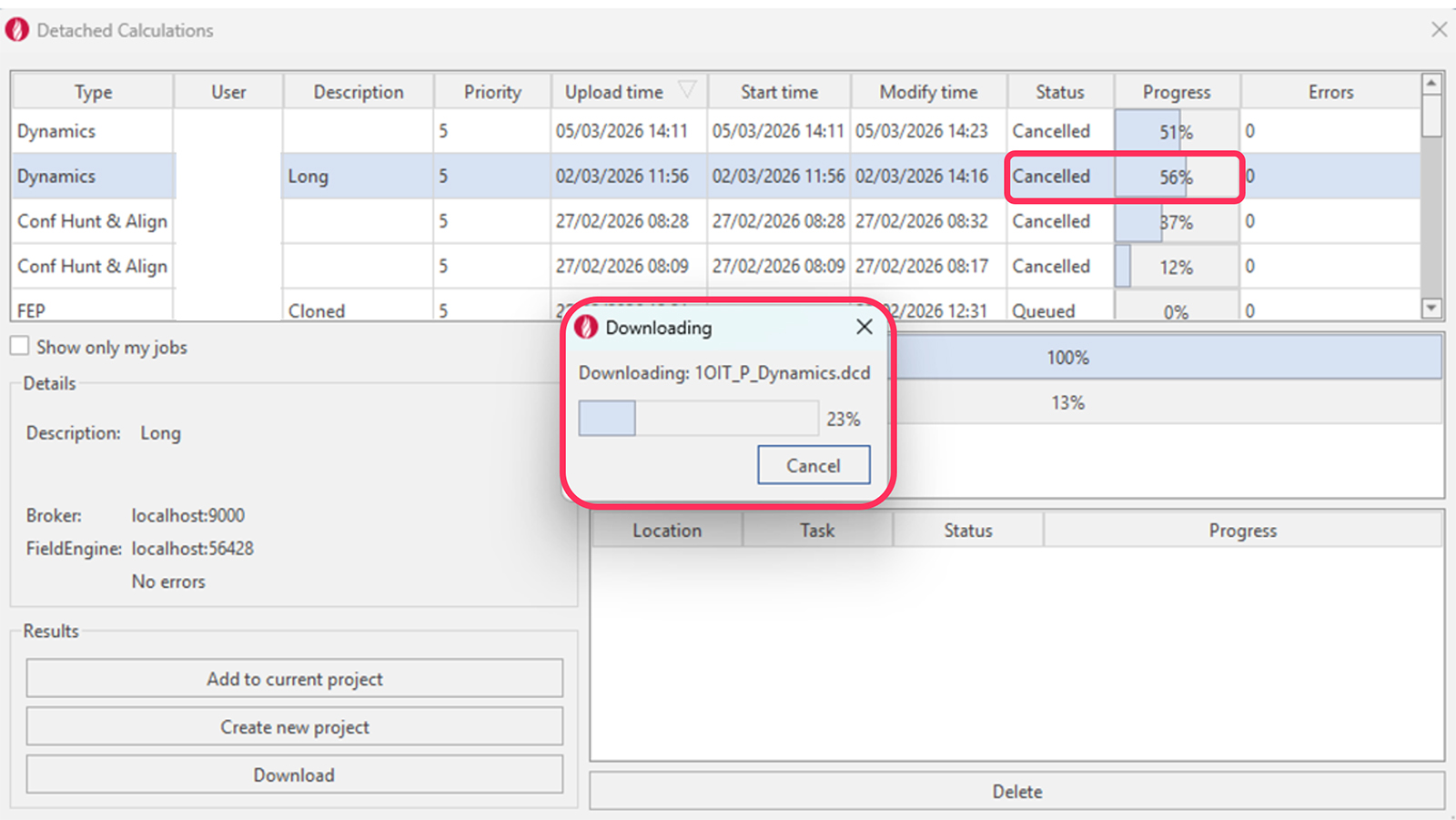
图 10. Flare V12/Cresset Engine Broker 3.6 中的分离式计算。
这些增强功能对于计算密集型工作流程(如长时间的分子动力学模拟)尤其有价值。例如,用户可以中断动力学模拟以评估体系稳定性、检查中间轨迹,或验证体系是否按预期运行。这降低了在可能需要调整的计算上投入大量计算时间的风险,并支持更具迭代性和响应性的模拟工作方法。
Flare V12 还通过 MolGenAI 为生成化学带来了分离式计算,增强了该方法的可用性;同时动力学和 FEP 支持创建自定义参数,使您能够以最适合分子体系的参数运行长时间计算。
借助 Flare V12 加速您的研究
Flare V12 强化了 FEP 在该平台中的作用,同时将其适用性扩展到更复杂的体系和变换。这些改进旨在帮助研究人员更高效地生成可靠预测,并支持在更广泛的药物发现挑战中做出决策。
请立即联系我们申请试用版,获取 Flare 最新功能的访问权限。我们的专家团队将指导您完成安装和设置,而我们丰富的教程库(涵盖从基本工作流程到高级方法)将确保顺利上手。借助 Flare V12,您将能够更快行动、更深挖掘,设计出最重要的分子。
联系我们: 020-38261356 info@molcalox.com
文献
- J. Zou, Z. Li, S. Liu, C. Peng, D. Fang, X. Wan, Z. Lin, T-S. Lee, D. P. Raleigh, M. Yang, C. Simmerling, Scaffold hopping transformations using auxiliary restraints for calculating accurate relative binding free energies. Journal of Chemical Theory and Computation, 2021, 17, 6, 3710–3726
- L. Wang, B. J. Berne, and R. A. Friesner, On achieving high accuracy and reliability in the calculation of relative protein–ligand binding affinities. PNAS, 2012, 109, 1937–1942
- G. A. Ross, H. E. Bruce Macdonald, C. Cave-Ayland, A. I. Cabedo Martinez, and J. W. Essex, Replica-exchange and standard state binding free energies with grand canonical Monte Carlo. Journal of Chemical Theory and Computation, 2017, 13, 6373–6381
- P. Atkins, J. dePaula, J. Keeler, Atkins Physical Chemistry 11th Edition, Oxford University Press, 2018
- https://github.com/openforcefield/ptm_prototype/releases/tag/v0.0.1a1


