
摘要:当前的分子对接评估通常侧重于几何精度(如RMSD),但可能忽视了结合模式的物理真实性。本研究通过深入分析POSEX基准测试中的一个典型算例——配体YI8(化合物7)在IPMK激酶中的交叉对接,发现其结论存在局限性。该算例中,人工智能(AI)方法报告的“成功”与基于物理方法的“失败”,均源于测试流程忽略了一个关键的保守水分子(Water 1),该水分子介导了核心的氢键网络。本研究证明,通过采用“闪烁水”(Flickering Waters)对接策略——在对接过程中动态考虑关键水分子的取舍,并在完整的物理力场下对初始对接构象进行松弛优化,能够成功重建出同时满足几何精度(RMSD从1.213降至0.684 Å)与物理真实性的结合模式。这项工作凸显了在分子对接评估与实践中确保“物理完整性”的重要性,并对当前过度依赖单一几何指标的评价范式提出了反思。
前言
在一项关于人工智能(AI)与传统基于物理的分子对接方法在交叉对接(Cross-Docking)任务中的性能对比研究(POSEX)中1,配体YI8(图1,化合物7)从其共晶结构PDB 8V71经结构比对映射至PDB 8V6Y蛋白的结合口袋时,与蛋白残基产生了显著的空间位阻(见原研究Figure S12)。具体而言,配体在其原生共晶结构(PDB 8V71)中的最优结合位置,在PDB 8V6Y的蛋白构象下被氨基酸侧链占据,导致了严重的立体冲突。该算例被列为AI方法显著优于基于物理方法的典型例证:研究中所有基于物理力场的对接算法均未能给出可靠的配体结合构象(预测构象与晶体结构间的均方根偏差\(RMSD≥2A˚\)),而AI对接方法SurfDock与AI共折叠方法AlphaFold3则被报告可精准预测其天然结合模式。
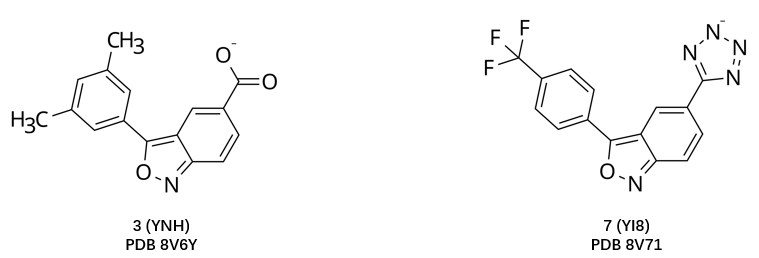
图1. IPMK激酶抑制剂3与7的化学结构式
通过分析化合物3(PDB 8V6Y的共晶配体)及化合物7(PDB 8V71的共晶配体)与IPMK激酶结构域的共晶结构(图2),可以发现一个保守的水分子(图中红色高亮标示的Water 1)发挥着关键作用2。该水分子介导了抑制剂羧基(化合物3)或四氮唑基团(化合物7)与激酶残基Glu86、Tyr90及Asp385之间形成的一个氢键网络。在POSEX的对比研究1中,由于计算流程忽略了对此关键水分子的考虑,其结果值得商榷:AI方法所“重现”的可能仅是重原子几何层面的近似(表现为较低的RMSD值),而非具有完整物理意义的结合模式(缺失了水介导的氢键网络);而传统基于物理的对接方法,则因在不完整的物理系统(无水环境)中进行计算,其“失败”并不能反映其在完备物理条件下真实的能力。
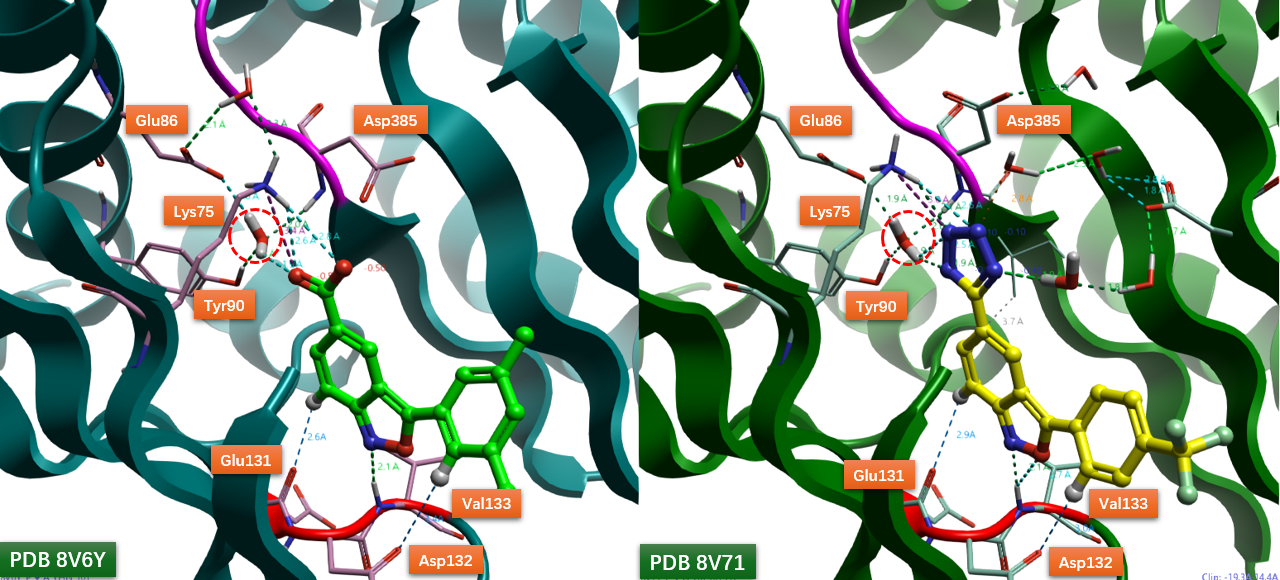
图2. IPMK激酶抑制剂3与7的结合模式。飘带图:IPMK激酶结构域;球棍模型:左侧为化合物3,右侧为化合物7。红色圆圈高亮处为保守水Water 12。
那么,在分子对接中,如何在未预先知晓关键水分子信息的情况下,仍能获得具有物理意义的结果呢?本文将介绍一种基于“闪烁水分子”(Flickering Waters)处理的对接策略3,并以Flare Docking软件4的实现为例。闪烁水分子特指蛋白质活性位点中那些在物理上可能被配体置换,也可能作为结合界面一部分的关键水分子。在对接过程中,将这些水分子标记为“闪烁”状态,算法将在采样与评分阶段动态决定是保留该水分子(作为受体的一部分参与相互作用)还是将其排除(由配体原子直接占据其位置),从而确保最终得到的结合模式在能量和相互作用上均更具物理真实性。
方法与结果
蛋白结构准备
在Flare (Version 11)中,下载化合物3、7与IPMK激酶结构域的共晶结构PDB 8V6Y与8V71。使用 Protein Prep 工具进行标准化预处理,包括添加氢原子、优化氢键网络、消除原子冲突及分配最佳质子化状态,截断的蛋白质链末端进行了封端处理。准备完毕,目视检查结合位点处蛋白与配体结构的正确性,并确保化合物7的四氮唑环处于去质子化状态。
蛋白3D结构叠合
使用Flare | Sequences | Align对所有的蛋白进行序列比对,然后再用Superimpose将准备好的PDB 8V71结构叠合到PDB 8V6Y上,结果如图3所示:化合物3、7的hinge结合骨架(苯并异噁唑)片段重原子几乎完全重合;与保守水Water 1结合的羧基与四氮唑片段也在3D上完全匹配。
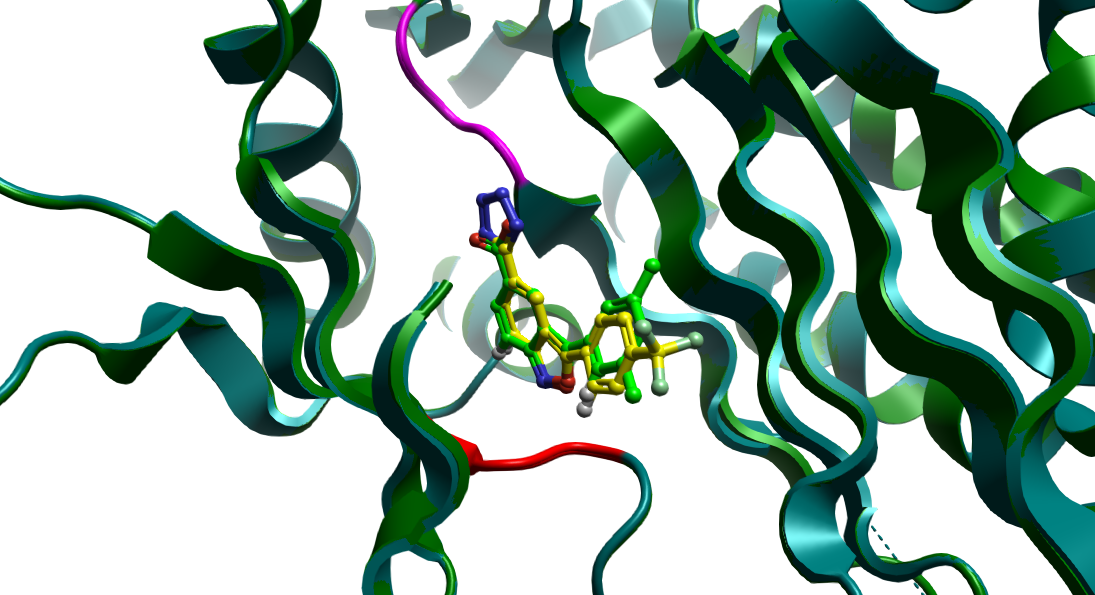
图3. IPMK激酶抑制剂3与7结合构象的叠合比较。绿色球棍:化合物3,黄色球棍:化合物7;细线:IPMK激酶结构域。
从构象上看,化合物3与7的主要区别是苯环-异噁唑可旋转键(见图4所示高亮处),化合物3的两面角Φ= -37 °而化合物7对应对两面角Φ= -28 °,虽然这个角度差异并不大,但是导致两个苯环重原子并不完全重合。
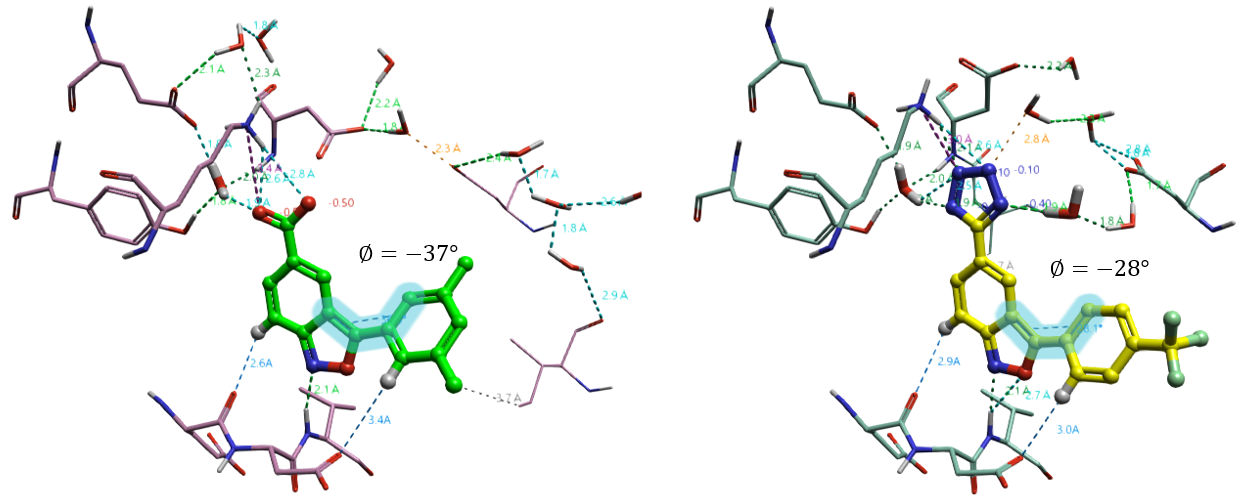
图4. IPMK激酶抑制剂3与7结合构象的叠合比较。绿色球棍:化合物3,黄色球棍:化合物7;细线:IPMK激酶结构域。
化合物7的结构准备
在Flare里,将化合物3、7分别从各种的复合物结构里提取到配体表单。接下来,将从SMILES开始为化合物7准备一个用于cross-docking的3D结构,流程如图7所示:在配体表单里,选择化合物7,Copy as SMILES;然后将SMILES复制到Create Ligand From SMILES;最后用Ligand Prep进行结构准备,生成3D结构。最后检查结构,有必要的情况下编辑四氮唑环,以确保其Formal Charge = -1。
图7. 化合物7的结构准备
闪烁水的分子对接实验
在将化合物7交叉对接到化合物3的结合位点里之前,需要先设置Water 1为闪烁水。在蛋白表单里选择准备好的8V6Y结构,为了设置方便,在3D视窗里选择Water 1,然后在蛋白表单里将该水clone一个到新的Chain B。然后隐藏Chain A的Water链,而仅展示蛋白与克隆到Chain B的Water 1。在3D视窗中右键点击Water 1,然后在菜单中选择“Mark as Docking Flickering Waters”,如图8所示。设置成功,你可以在3D视窗上看到Water 1旁边出现一个“Flickering Water”的标识。
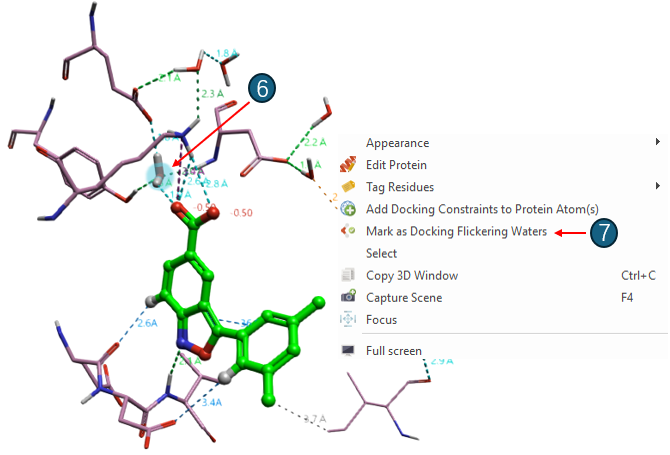
图8. 设置闪烁水
在配体表单单击选中之前从SMILES开始准备的化合物7,然后点击3D Pose | Dock | Normal打开分子对接计算(Docking Calculation)面板,如图9所示。
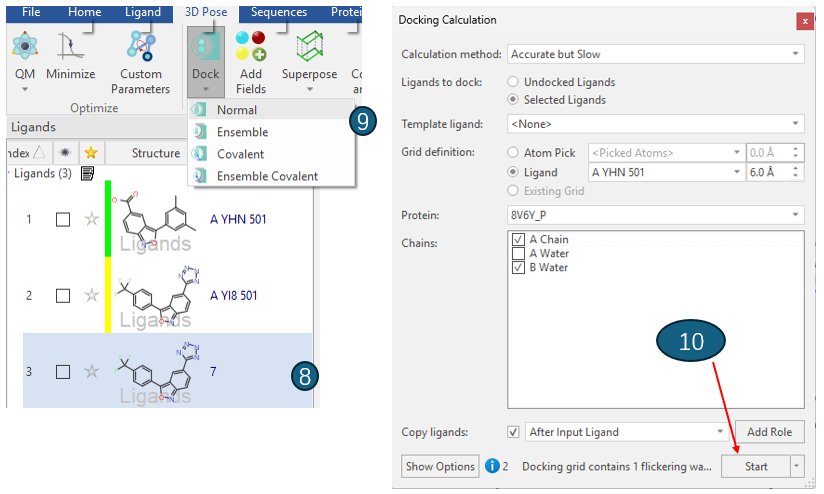
图9. 化合物7的对接实验
对接实验参数如下:
- Calculation Method: Very Accurate but Slow
- Ligands to Dock: Select Ligands
- GRID definition: A YHN 501
- Protein: 8V6Y_P, A Chain and B Water
其中B Water仅包含一个Flickering Water。
再这次对接实验中,共生成了9个结合模式。以PDB 8V71的共晶配体(天然结合模式)为参比,最佳结合模式的RSMD=1.213 Å,如图10所示。比较天然结合模式与对接结合模式,可以发现,天然结合模式的四氮唑环与8V6Y的蛋白发生立体碰撞,对接结合模式为了避开这个碰撞,发生的重原子坐标偏移,即便如此,但是关键相互作用得以维持存在。
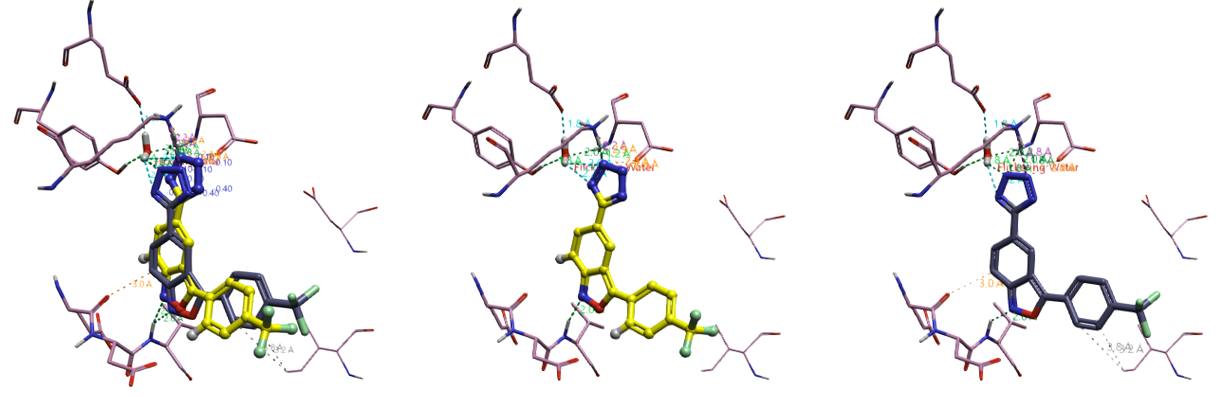
图10. 化合物7的交叉对接实验结果。黄色球棍:化合物7的天然结合模式(PDB 8V71);棕色棍棒:化合物7的对接到PDB 8V6Y的结合模式。左:对接与天然的结合构象叠合比较,RMSD = 1.213 Å。
结合模式的松弛处理
为优化对接获得的复合物结构,参照POSEX研究1中提出的策略,对初始构象进行了基于分子力场的能量最小化(松弛,Relaxation)处理。具体而言,我们将对接获得的复合物结构作为输入,在OpenMM引擎中执行能量最小化计算。计算中,蛋白质采用AMBER14-all力场,配体小分子采用OpenFF-2.2.1力场,水模型采用TIP3P。为在优化局部接触的同时保持蛋白质的整体骨架,我们对所有蛋白质Cα原子施加了力常数为2.5 kcal/mol/Ų的谐波势约束。能量最小化以梯度收敛容限0.24 kcal/mol/Å为停止标准。
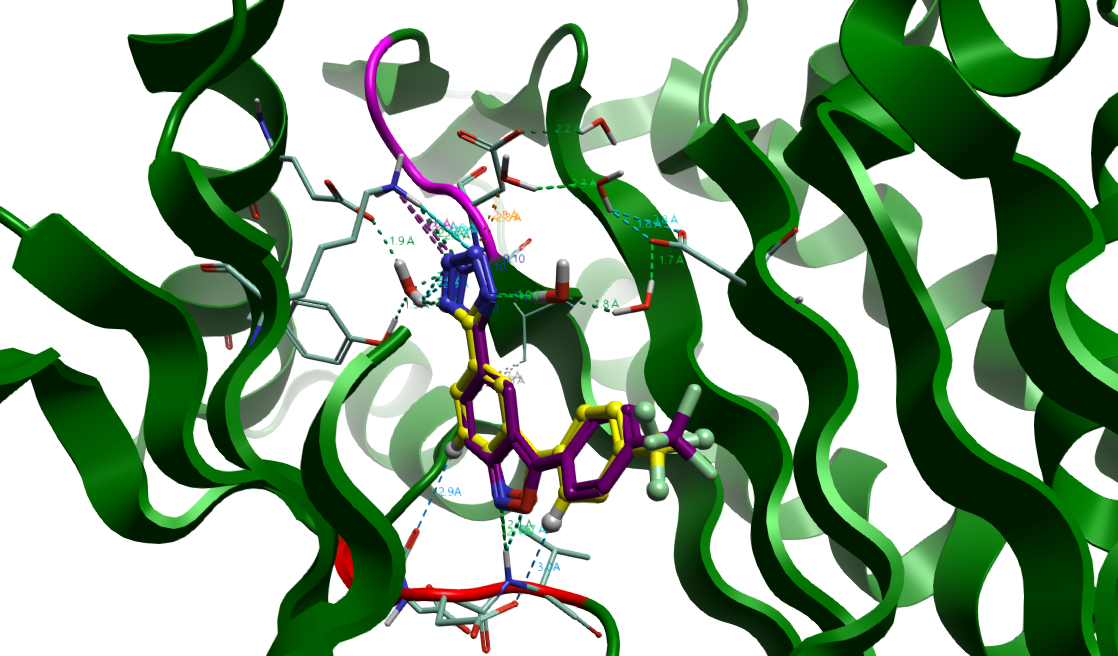
图11. 化合物7的交叉对接实验结果Relax处理。飘带:IPMK激酶结构域(PDB 8V71);黄色球棍:化合物7的天然结合模式(PDB 8V71);紫色棍棒:经Relaxed处理的化合物7交叉对接结合模式。Relax处理的对接结合模式与天然的结合构象叠合比较,RMSD = 0.684 Å。
经松弛优化后的复合物结构与化合物7-IPMK共晶结构(PDB 8V71)叠合结果如图11所示。松弛后的化合物7构象(紫色棍棒)与天然结合构象(黄色球棍)高度一致,其重原子RMSD为0.684 Å。除三氟甲基之外,配体其余重原子位置几乎完全重合。该结果验证了松弛处理对提升对接构象几何精度的有效性:经优化后,构象偏差(RMSD)从松弛前的1.213 Å显著降低至0.684 Å,这与POSEX研究中“Relax改善对接结果”的结论相一致。更重要的是,保守水Water 1参与的相互作用(未展示,请从附件Flare项目文件查看)也得以重现,还原了化合物7物理真实的天然结合模式,而这是AI对接与共折叠方法所缺失的。
闪烁水对接应用算例
BTK抑制剂的水分子替换
闪烁水分子是指蛋白质活性位点中那些在对接实验中可被“开启”或“关闭”的水分子。尤其是桥接水分子,可能被某些对接的配体置换,但不会被其他配体置换。例如,在下图中,PDB 4ZLZ共晶配体(左图)的吡啶通过水分子954的桥接,与蛋白质的活性位点形成水介导的相互作用。而在PDB 4Z3V(右图)中共晶配体的吲唑片段则直接与BTK的活性位点相互作用,取代了桥接水分子。
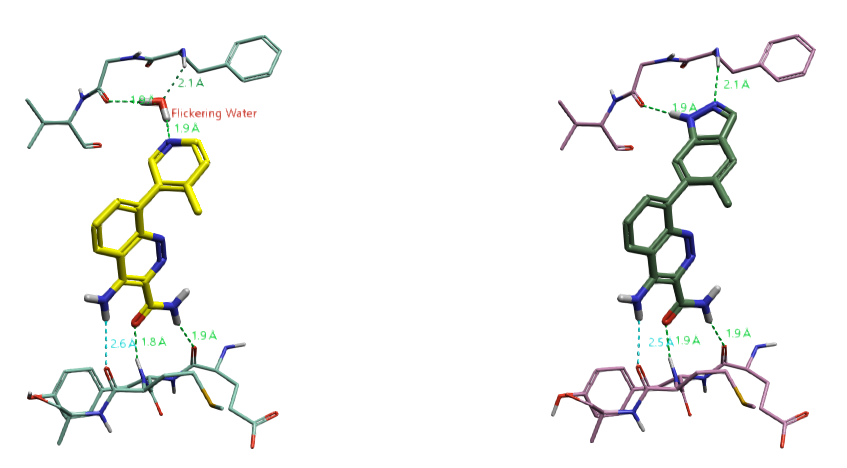
图12. BTK抑制剂的共晶结构。左:PDB 4ZLZ;右:PDB 4Z3V。
JAK1抑制剂:水的取代与否影响靶标选择性
Tofacitinib与JAK1的共晶结构PDB 3EYG中,如图13所示,HOH1198与Gly1020的主链羰基以及Asp1021的侧链羧酸发生形成氢键相互作用网络,还与Gatekeeper Met956形成O/S超共轭效应。
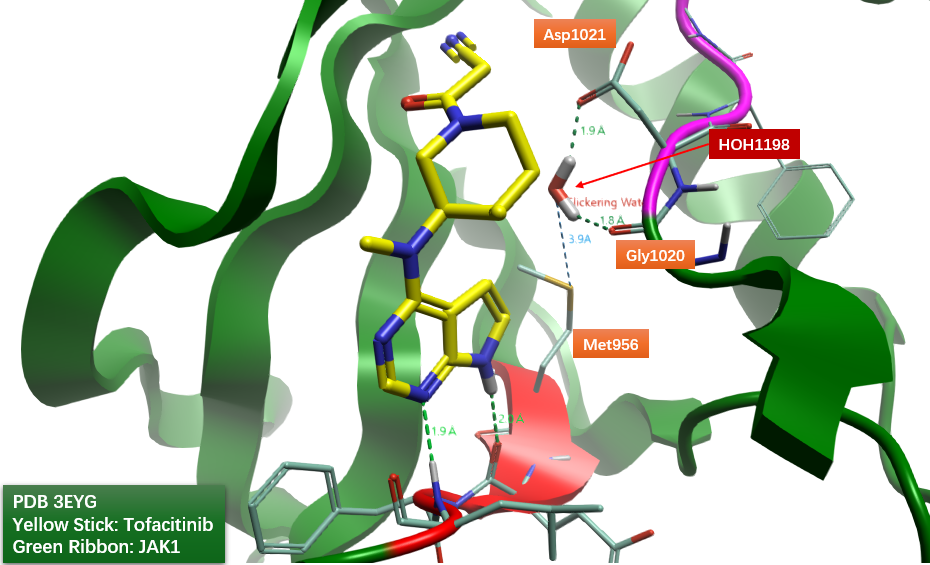
图13. JAK1抑制剂Tofacitinib的共晶结构PDB 3EYG。飘带图:JAK1;红色飘带:激酶Hinge区;粉色飘带:DFG片段;黄色棍棒:Tofacitinib。
水分子1198的存在导致JAK1激酶ATP结合口袋出现不寻常的构象,它阻断了通向”后口袋(back pocket)”的通道。这一特性可能是托法替尼相较于许多其他激酶具有相对较高选择性的原因之一。Ritzén等人5的研究表明,当设计的抑制剂能够置换该水分子、移动守门残基Met956、并延伸到激酶后口袋中,与托法替尼相比,这些抑制剂对其他激酶的选择性要低得多。因此,在对接计算时该水的保留与否至关重要,可以通过闪烁水对接来提高对接成功率。
结论
本文通过深入剖析一个典型的交叉对接算例,揭示了当前分子对接评估中一个容易被忽视的关键问题:物理系统的完整性。POSEX研究中关于AI方法与物理方法对比的结论,由于在测试设定中忽略了对结合模式至关重要的保守水分子,可能对两种范式的真实能力做出了误判。
我们的工作表明:
- “闪烁水”策略的有效性:在对接中显式考虑并动态处理关键水分子,是连接几何精度与物理真实性的有效桥梁。它允许算法在更完备的物理环境中进行采样和评分,从而能够复现出像Water 1介导的氢键网络这类关键相互作用。
- 物理方法在公平条件下的潜力:当在一个物理描述完备的体系(包含关键溶剂效应)中运行时,基于物理力场的对接与松弛方法能够生成不仅几何上高度精确(\(RMSD < 0.7 Å\)),而且相互作用网络物理真实的结合模式,这为后续的理性药物设计提供了可靠起点。
- 对评估范式的反思:单一的几何指标(如RMSD)不足以评价结合模式的真实性。一个“低RMSD”的构象如果缺失了关键性的物理相互作用,其科学价值与指导意义将大打折扣。未来的对接方法评估应纳入对关键相互作用重现度的考量,并确保测试系统本身的物理完整性。
综上所述,分子对接研究应从追求单纯的“几何吻合”迈向追求“物理真实”。本研究倡导的“闪烁水”处理及对物理完整性的重视,不仅纠正了特定案例的结论,更为旨在获得可解释、可转化生物物理洞察的计算药物发现实践,提供了切实可行的策略与反思。
附件
化合物7-IPMK共晶结构、化合物7交叉对接结果及其Relax结果的Flare项目文件:Compound-7-Cross-docking-and-Relax.flr 提取码: w518
在自己的项目中使用Flare的闪烁水对接
最新版Flare V11 引入了强大的新科学能力、更快的工作流和增强的易用性,为您提供高效、精准探索复杂体系的工具。立即联系我们申请试用,体验 Flare 的最新功能。我们的专家团队将指导您完成安装与设置,同时我们丰富的教程库(从基础工作流到高级方法)确保您顺利上手。借助 Flare V11,您将能够更快推进研究、深入洞察机制,并设计出最具价值的分子。
联系我们: 020-38261356 info@molcalox.com
文献
- Yize Jiang, Xinze Li, Yuanyuan Zhang, et al. “PoseX: AI Defeats Physics Approaches on Protein-Ligand Cross Docking.” arXiv:2505.01700. Preprint, arXiv, April 26, 2026. https://doi.org/10.48550/arXiv.2505.01700.
- Wang, H.; Shears, S. B.; Blind, R. D. Structural Rationalization of IPMK Inhibitor Potency. J. Med. Chem. 2025, 68 (22), 24316–24325. https://doi.org/10.1021/acs.jmedchem.5c02314.
- Protein-Ligand Docking. https://cresset-group.com/method/protein-ligand-docking
- Flare (Version 11). http://www.cresset-group.com/software/flare
- Ritzén, A.; Sørensen, M. D.; Dack, K. N.; Greve, D. R.; Jerre, A.; Carnerup, M. A.; Rytved, K. A.; Bagger-Bahnsen, J. Fragment-Based Discovery of 6-Arylindazole JAK Inhibitors. ACS Med. Chem. Lett. 2016, 7 (6), 641–646. https://doi.org/10.1021/acsmedchemlett.6b00087.


