
摘要:自由能微扰(FEP)计算在药物发现中可用于预测配体与靶蛋白的结合亲和力,帮助优先选择化合物进行合成和测试。本研究旨在评估 Flare FEP 在生产模式下对 TYK2 激酶抑制剂进行前瞻性预测的能力。首先以 16 个已知活性的 TYK2 基准化合物进行基准实验,验证 Flare FEP 的预测准确性(r² = 0.8,MUE = 0.50);随后,对 18 个来自 Liang et al.(2013)的铰链结合变体进行前瞻性盲预测,预测时仅使用一个已知活性化合物(ejm_46)作为锚点,其余 17 个配体的活性数据在预测过程中被完全屏蔽。自动构建的 FEP 扰动网络包含 19 个化合物、27 条双向链接(共 54 个微扰),预计计算耗时为 265 GPU 小时。预测的最佳结合配体为 Liang_6(ΔG = -13.1 kcal/mol),最差为 Liang_15(ΔG = -9.9 kcal/mol);预测前十名中,有 8 个配体在实验中被确认为强结合剂,且预测最差的配体(Liang_15)与实验结果一致。但 Liang_6 和 Liang_11 被明显高估(偏差分别为 2.6 和 1.1 kcal/mol),导致整体 Pearson R² 仅为 0.21;数据集的整体动态范围较小(2.5 kcal/mol),低于 FEP 基准实验的建议值(3.0 kcal/mol),预测的平均无符号误差(MUE)为 0.59 kcal/mol。尽管在绝对数值预测上精度有限(R² 较低),Flare FEP 在生产模式下成功识别了数据集中的最佳和最差结合配体,在前十名预测中正确捕获了 8 个实验活性较强的化合物;结果表明,FEP 可作为先导化合物优化中的辅助排序工具,帮助优先选择合成目标、减少低活性化合物的合成投入,但应注意其在结合能绝对值预测上的局限性。
Lauren Nelson/March 24, 2026. A new prospective study of TYK2 with Flare FEP™.
引言
自由能微扰(FEP)计算可精准对项目内配体进行亲和力排序,以此加快配体设计进程。在前期研究\(^{1}\)中,我们在基于文献公开的FEP基准数据集验证中得到了具有竞争力的计算结果。本文介绍一项新的案例研究:通过盲试实验,前瞻性预测一系列化合物与TYK2的结合亲和力,以模拟实际药物研发项目筛选优先候选分子的工作流程。
TYK2属于Janus激酶(JAK)家族,该家族一共包含4个成员:JAK1、JAK2、JAK3以及TYK2;每种激酶分别结合特定受体进而启动胞内信号通路\(^{2}\)。TYK2可与多种细胞因子受体相结合;抑制TYK2激酶活性是银屑病、克罗恩病等自身免疫性及炎症性肠病的重要治疗研发思路\(^{3,4}\)。
Liang等人在2013年发表的研究\(^{2,3}\)报道了一组同系物的配体分子,用于探究小分子在TYK2蛋白活性口袋两处关键结合区域:铰链区以及连接区\(^{2}\)。该篇文章中的16个配体现已成为TYK2靶点研究中常用的FEP基准数据集\(^{1,5}\)。这批化合物来自同一研发项目,均含有4‑氨基吡啶‑苯甲酰胺母核结构,因此我们可以基于这套基准体系,对更多化合物开展前瞻性FEP计算工作。本研究选取该文献中针对连接区改造的18个衍生物\(^{3}\)(即在上方苯环C4位进行R基团修饰,见图1‑B),采用Flare FEP软件在生产模式下完成本次盲预测试验(blind production run)。
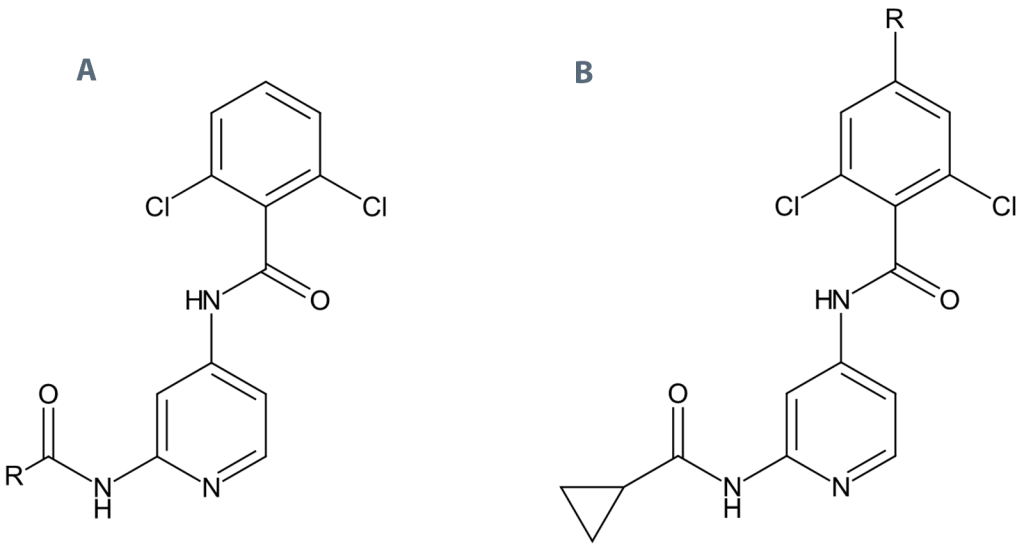
图1. Liang 数据集\(^{2,3}\)中配体的公共 4-氨基吡啶苯甲酰胺母核,图中标注了 R 基团的修饰位点:(A)针对 TYK2 活性口袋的铰链结合区;(B)针对连接区。在图 1-B 中,当 R 为氢原子时,该分子即为 PDB 编号 4GIH 的晶体配体 0X5,也就是 Liang 等人\(^{2}\)文章中的化合物 3。图 1-A 中的分子为 Flare FEP 基准测试所用的 16 个配体,图 1-B 中的 18 个配体则用于生产模式下的盲预测计算(blind production run)。
方法
TYK2基准化合物的FEP实验
在此实例中,我们已成功运行了 TYK2 体系的基准实验\(^{1}\)。以下我将简要回顾准备步骤:
我们首先仔细准备蛋白质(PDB:4GIH),使用Flare中的自动化蛋白准备工具(Protein Prep),随后对活性位点进行细致检查。然后,我们采用分子动力学(MD)与巨正则非平衡候选蒙特卡洛(GCNCMC)方法,以确保模型能够捕获生物活性配体-蛋白质的结合模式。最后,我们使用带有晶体学配体(0X5)的3D-RISM水稳定性分析\(^{6}\)添加水分子位点,使体系在进行基准FEP计算前具有适当的水合状态。关于在Flare FEP基准研究之前推荐使用的蛋白质准备步骤的更详细说明,请参考我们之前的案例:快速准确的先导化合物发现过程:CDK9抑制剂 和 膜靶点上的FEP:捕获P2Y1 GPCR复合物中的脂质暴露结合。在此,我将简要回顾蛋白质准备及后续的FEP基准测试过程:
使用已准备好的蛋白质,使用“Conformational Hunt and Alignment”工具将包含16个配体的基准数据集\(^{1}\)叠合到共晶的配体上,以获得尽可能紧密的叠合(图2,A)。然后将这些配体构象和准备好的蛋白质用于评估Flare FEP预测与实验结合亲和力之间的性能。
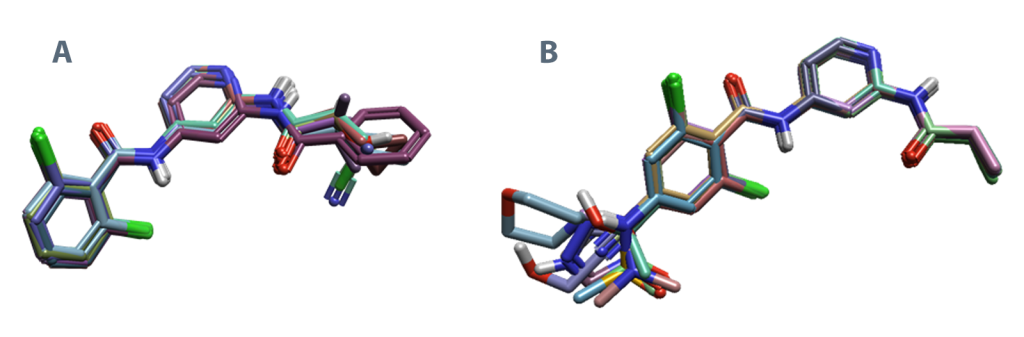
图2.(A)基准实验中叠合后的配体(16个配体)\(^{1}\) 与 (B)生产性FEP盲测预测实验中的18个配体\(^{2}\)的比较。配体使用Flare中的Conformational Hunt and Alignment子结构方法进行叠合。同样,(A)探索了铰链结合区,而(B)则研究了TYK2活性位点的连接区。
通过在基准研究前进行这些细致的准备工作,我们获得了比 Wang 等人\(^{5}\)更好的预测亲和力及统计结果(r² = 0.8,MUE = 0.50),从而免去了后续对有问题的扰动进行修改和排查的麻烦。该基准测试结果如图 3 所示。
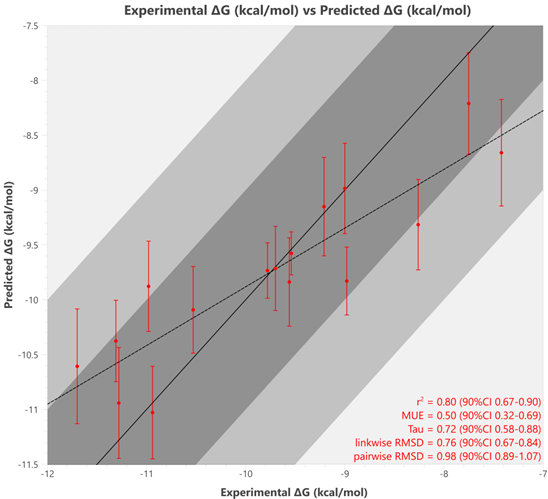
图3. 16 个 TYK2 基准化合物的 Flare FEP 预测 ΔG 值(kcal/mol)与实验 ΔG 值\(^{1,5}\) 的对比图,误差线由 Flare FEP 计算得出。
对未知铰链结合设计的预测
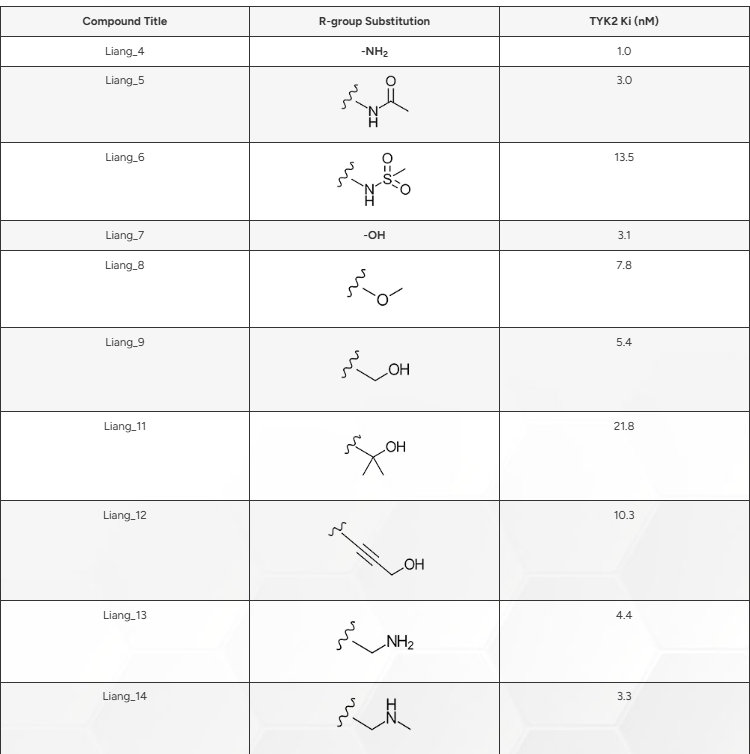
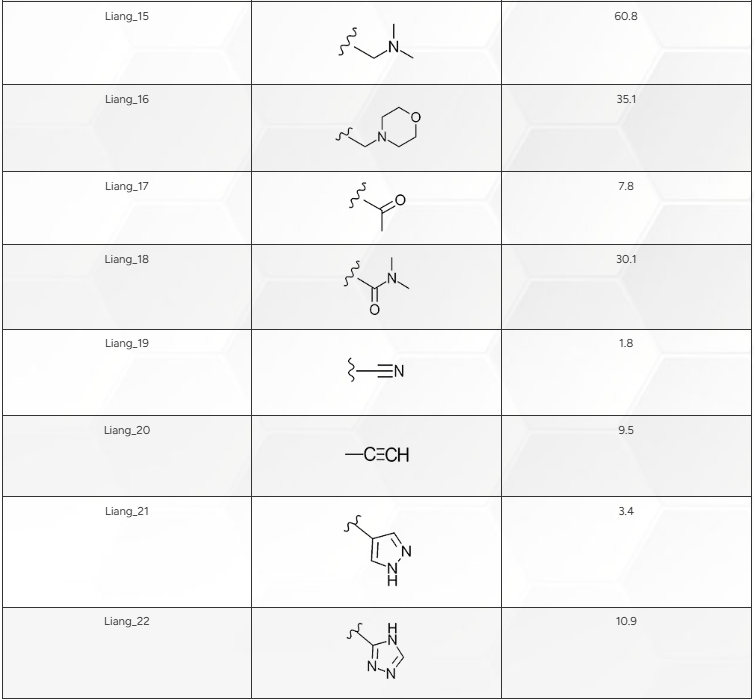
在对基准实验的性能建立信心后,我们从铰链结合变体\(^{2}\)中确定了18个配体。这些配体均含有基准配体集的母核结构,但在母核苯环的C-4位引入了不同的取代基(见图1,B)。表1详细列出了各取代基及其对应的结合亲和力,不过这些亲和力数据直到评估预测结果时才被使用。值得注意的是,这些配体的活性范围为1.0至60.8 nM(pIC50为9.0至7.2)。
表1. 图1所示结构苯环C4位上的修饰及其相关Ki值(nM),由Liang等人\(^{2}\)测定。
将16个TYK2基准化合物(已知活性,图2,A)与18个来自铰链结合数据集的配体(输入时无活性数据,图2,B)一同输入后,系统自动创建了生产性FEP扰动图(production FEP perturbation map)。该图仅包含一个已知活性的化合物——来自基准系列的ejm_46,其中R = H(苯环C-4位),见图1,B。
该网络在19个化合物之间包含27条链接(links),每条链接沿正反两个方向运行,共产生54个微扰( perturbations),用以衡量每条链接的滞后性(hysteresis)。由于未知化合物与网络中所选已知化合物在结构上较为相似,各链接的打分良好,无需额外添加中间体链接。不过,其打分范围仍然较宽(0.55至0.95),说明其中部分化学变换并不简单。整个FEP网络的计算预计需要265个GPU小时。
结果
如开头所述,我们通过这次生产运行对18个”新”铰链结合变体进行排名,以优先选择用于合成的设计。预测的最佳结合配体为Liang_6(ΔG = -13.1 kcal/mol,±1.3 kcal/mol),最差为Liang_15(ΔG = -9.9 kcal/mol,±0.5 kcal/mol)。RBFE计算的目标误差为2 kcal/mol以内。本数据集中仅Liang_16的误差(±2.3 kcal/mol)超出该范围,其余配体的预测误差均在2 kcal/mol以内。预测误差越小,结果越可靠。前十名预测结合配体(ΔG最低)在表2中以粗体列出。我们将\(ΔG ≤ -11.0 kcal/mol\)作为筛选标准,共选出10个可推进的配体。该阈值参考了已知活性化合物ejm_46(基准系列,ΔG = -11.3 kcal/mol)的结合能。值得一提的是,Flare FEP生产模式中的已知活性化合物由软件根据与未知配体的链接质量自动选择,而非依据活性。
其余8个配体(我们不会将其推进测试)的预测结合亲和力在-9.9到-10.8 kcal/mol之间。在实际项目中,如果某个配体有助于支持解决其他问题(如溶解度或ADME性质)的假设,用户可能希望从这组”较差”结合配体中选取一个推进。从表2以及迄今为止引用的预测结合亲和力范围可以看出,数据集中间的预测存在重叠。有7个化合物落在-10.6到-11.2 kcal/mol的狭窄0.6 kcal/mol范围内(Liang_8、9、12、13、14、19和20)。在这种情况下,如果用户希望从数据集的这一部分选取化合物推进,他们可以评估自己的假设,选择最能解决项目其他问题(如溶解度)的化合物,因为这些配体的误差值与我们应用的任意ΔG截止值存在重叠。
表2. Flare FEP预测的铰链结合系列的结合能和排名顺序。同时列出了预测活性的相关误差(kcal/mol)。前十名结合配体(最低ΔG值)以粗体突出显示。
| 配体名称 | 预测ΔG (kcal/mol) | 预测排名 | 预测活性误差ΔG (kcal/mol) |
|---|---|---|---|
| Liang_6 | -13.1 | 1 | 1.3 |
| Liang_5 | -12.4 | 2 | 1.4 |
| Liang_21 | -12.4 | 2 | 1.3 |
| Liang_4 | -12.2 | 4 | 1.1 |
| Liang_7 | -11.6 | 5 | 0.6 |
| Liang_11 | -11.5 | 5 | 0.5 |
| Liang_9 | -11.2 | 7 | 0.5 |
| Liang_8 | -11.1 | 8 | 0.5 |
| Liang_13 | -11.0 | 9 | 0.4 |
| Liang_19 | -11.0 | 9 | 0.8 |
| Liang_14 | -10.8 | 11 | 0.6 |
| Liang_12 | -10.8 | 11 | 0.7 |
| Liang_20 | -10.6 | 13 | 0.9 |
| Liang_16 | -10.4 | 14 | 2.3 |
| Liang_18 | -10.2 | 15 | 0.6 |
| Liang_22 | -10.2 | 15 | 1.5 |
| Liang_17 | -10.1 | 17 | 0.5 |
| Liang_15 | -9.9 | 18 | 0.5 |
现在我们可以比较配体在实验ΔG值与预测ΔG值方面的排名顺序(表3)。对于预测的前十名最佳结合配体,实验排名与预测排名基本一致。尽管存在更困难的扰动(添加五个重原子以生长一个吡唑环),Liang_21仍然被很好地预测并排名靠前,与实验排名相符,成为数据集中较好的结合配体。然而,有两个化合物被过度预测了:Liang_6高出2.6 kcal/mol,Liang_11高出1.1 kcal/mol,因此排名高于实验值。如前所述,RBFE计算中预期误差为2 kcal/mol或更小,因此Liang_11并未超出我们的预期范围。然而,由于活性范围有限,数据集中间的化合物在某些情况下仅相差0.2 kcal/mol。因此,由于Liang_11被高估了1.1 kcal/mol,使其从实验排名的第15位上升到了预测数据的第5位。Liang_6包含一个磺酰基添加,而Liang_11则是向叔丁基的扰动,两者都是较为复杂的转化,我们可以进一步详细考虑。
表3. Flare FEP预测的前十名结合配体(最低ΔG值)与相应的实验值(nM)\(^{2}\)及同一配体的排名对比。同时列出了预测活性的相关误差(kcal/mol)。
| 配体名称 | TYK2 Ki (nM) | 实验ΔG (kcal/mol) | 实验排名 | 预测ΔG (kcal/mol) | 预测排名 | 预测活性误差ΔG (kcal/mol) |
|---|---|---|---|---|---|---|
| Liang_6 | 13.5 | -10.7 | 14 | -13.1 | 1 | 1.3 |
| Liang_5 | 3 | -11.6 | 3 | -12.4 | 2 | 1.4 |
| Liang_21 | 3.4 | -11.6 | 3 | -12.4 | 2 | 1.3 |
| Liang_4 | 1 | -12.3 | 1 | -12.2 | 4 | 1.1 |
| Liang_7 | 3.1 | -11.6 | 3 | -11.6 | 5 | 0.6 |
| Liang_11 | 21.8 | -10.4 | 15 | -11.5 | 5 | 0.5 |
| Liang_9 | 5.4 | -11.3 | 8 | -11.2 | 7 | 0.5 |
| Liang_8 | 7.8 | -11.1 | 9 | -11.1 | 8 | 0.5 |
| Liang_13 | 4.4 | -11.4 | 7 | -11.0 | 9 | 0.4 |
| Liang_19 | 1.8 | -11.9 | 2 | -11.0 | 9 | 0.8 |
含硫基团已知对FEP计算更具挑战性。力场与硫相关的问题包括:(i) 硫往往是不对称的,因此点电荷可能不足;(ii) 硫足够大,以至于对于CHNO等基团可接受的廉价QM方法在处理硫时可能会遇到困难;(iii) 如果力场试图对所有硫使用同一参数,这可能会引发问题,因为SO₂与SO和单独的S差异很大。这类困难通常表现为添加或移除硫的FEP循环中出现较大的滞后性。然而,在本例中,包含Liang_6的循环的滞后性是可接受的(±1.1 kcal/mol),Liang_6的估计误差为1.3 kcal/mol。但通过使用Flare FEP中的”收敛性”(Convergence)分析进一步研究,我们可以看出结合态在正反两个方向上都存在收敛不足的问题。因此,如果我们为与Liang_6化合物相关的链接添加一个”实例”(instance),我们将延长结合态的模拟时间(从4ns到8ns),以实现更好的收敛性,并有希望改善对该配体的预测。图4展示了一个示例。
改变FEP计算中选定链接设置的模拟时间
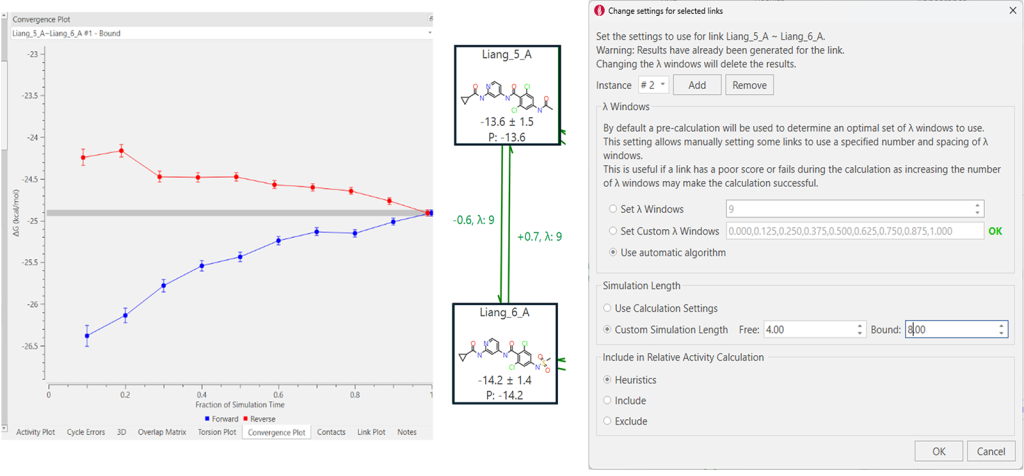
图4. 展示了一种简便方法:当FEP网络中的结合模拟收敛不理想时,只需添加一个实例,即可仅增加该支路(结合态)的模拟时间,而无需同时延长结合态和游离态两者的模拟时长。
总体而言,如果我们根据Flare FEP的排名发送预测的前十名结合配体(预测ΔG为-11 kcal/mol或更低),其中8个在实验中被证实具有活性,只有2个比预测的弱。值得注意的是,Flare FEP在前五名化合物中捕获了已知的最佳结合配体Liang_4,并且仅以微弱差距遗漏了一个化合物Liang_14(实验ΔG为-11.6 kcal/mol,预测ΔG为-10.8 kcal/mol),使其未能进入前十名。该配体在预测研究中排名第11,误差为±0.7 kcal/mol,因此是否将该化合物纳入进一步测试将由用户决定。重要的是,Flare FEP还正确预测了数据集中最差的结合配体Liang_15(ΔG_exp = -9.8 kcal/mol,ΔG_pred = -9.9 kcal/mol ±0.5 kcal/mol),证明我们可以减少不必要进入检测的弱结合配体的数量。
值得注意的是,前十名最佳结合配体之间的动态范围仅为1.2 kcal/mol,整个数据集的总体活性范围为2.5 kcal/mol,略低于建议值(3.0 kcal/mol)(如果这是一个基准系列的话)\(^{7}\)。在一个好的FEP研究中,你可以预期误差在1-2 kcal/mol之间,因此在创建基准时需要有大于此范围的动态范围。因此,尽管相关性(r²)较低,Flare FEP仍然正确预测了实验检测中前十名结合配体中的8个,误差在可接受的能量范围内。
我们回顾性地绘制了18个配体\(^{2}\)的实验ΔG值与Flare FEP生产运行预测ΔG值的对比图,如图5所示。
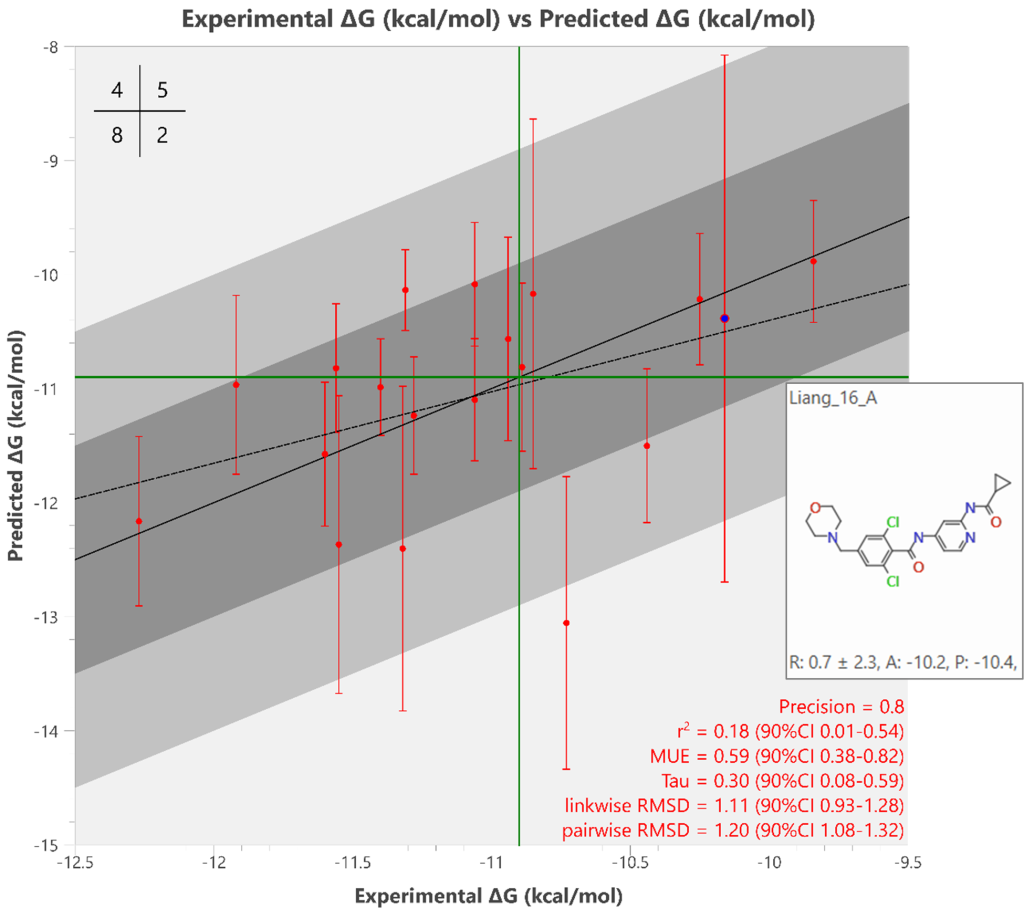
图5. Flare FEP 预测的 19 个化合物的 ΔG 值(kcal/mol)与实验 ΔG 值[2] 的对比图。图中包含由 Flare FEP 计算的误差线,以及一个精确度矩阵(左上角),其交叉点(绿色十字)设为 -10.9 kcal/mol,用于捕获 Flare FEP 排名前十的结合配体(其中 8 个落入该范围)。化合物 Liang_16 以二维结构形式展示,如同将鼠标悬停于活性图上的对应数据点时显示的效果。
尽管统计输出可能不显示预测ΔG值与实验ΔG值之间的强相关性,但数据集中最佳和最差结合配体的排名是正确的。将精确度设置为捕获Flare FEP排名前十名结合配体中的8个配体(-10.9 kcal/mol,见图5),左上角的混淆矩阵显示8个真阳性、4个假阴性(其中一个是已知活性的基准化合物ejm_46)、5个真阴性和2个假阳性,即获得真实结果的概率为80%。如前所述,动态范围的中间区域存在显著重叠,其中一些化合物具有较大的误差线(如配体Liang_16,图5)。较大的误差线(即大于2 kcal/mol)表明我们应对与该配体相关的预测信心较低。然而,图表两端(即最佳和最差结合配体)的大多数化合物预测良好,误差较低。平均无符号误差(MUE)非常出色,为0.59 kcal/mol。
故障排除
Flare FEP在基准模式和生产模式研究中提供多种工具来排查有问题的链接,包括添加链接实例的选项,无需重新运行整个FEP网络,从而节省计算时间。在27条链接中,只有一条未被Flare纳入结果,因为该链接的滞后性过高,且已为该问题配体Liang_16添加了其他链接实例。这个链接位于化合物Liang_9和Liang_16之间,分别从羟基团扰动到吗啉环,没有中间体,如图6所示。Flare FEP中的自动中间体生成器建议化合物Liang_13是最合适的中间体(其中R = CH₂NH₂),因此在网络生成时未包含第二个中间体。
Liang_16还具有较高的误差范围,为±2.5 kcal/mol。在这种情况下(即高误差和高滞后性),用户可能希望选取配体Liang_16(因为它可能非常活跃或完全不活跃)并通过实验测定,因为预测不够稳健,但该化合物可能对用户具有化学意义。然而,这显然取决于具体项目。
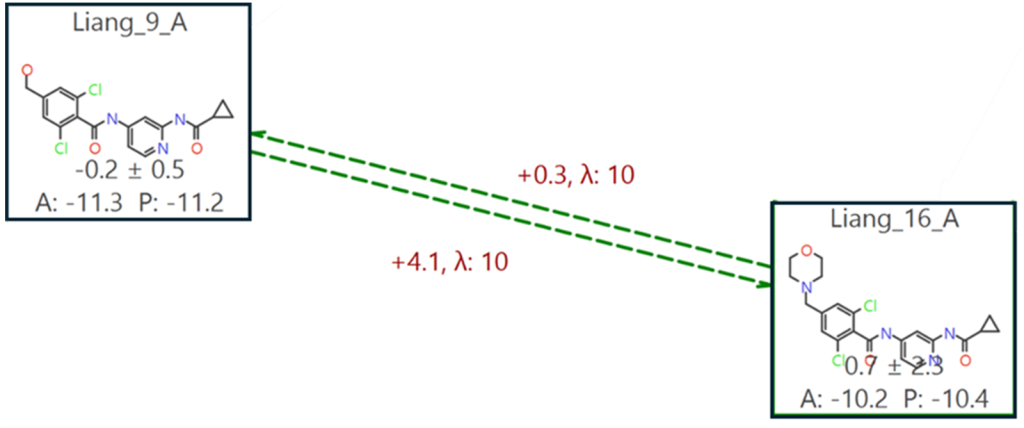
图6. 从Liang_9到Liang_16的扰动具有高滞后性,其中羟基需要扰动为吗啉环。
在Flare FEP中,您可以根据链接对总误差的贡献百分比为链接着色。这样,我们可以快速识别那些如果也表现出滞后性则可能受益于额外实例的链接,如图7中Liang_7和Liang_22之间的链接所示。
在本实验的情况下,我们选择不为扰动图中的任何链接添加更多实例,因为这是一个生产运行,而非基准研究(在基准研究中我们可能会花更多时间微调实验设置)。在这里,我们想确定生产模式是否能够使用基准测试中使用的参数(在本例中为默认设置,并在平衡过程中使用GCNCMC)对这些化合物提供”好或坏”的稳健预测。我们还可以使用Flare FEP中的”增强连接性”(Enhance Connectivity)工具,为图谱中较弱的区域添加更多可能的链接。
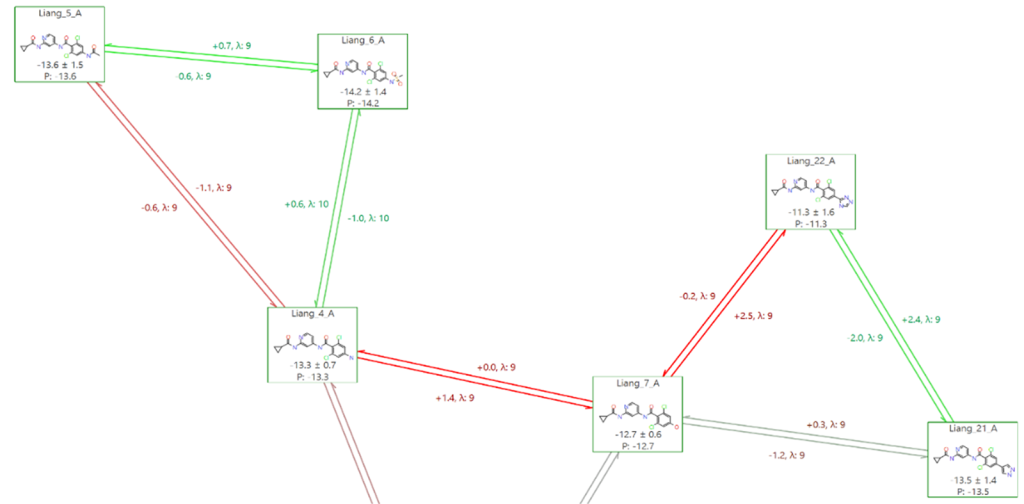
图7. Flare FEP 中的”按误差百分比为链接着色”功能:红色链接对总误差贡献最大,绿色链接贡献最小。该功能可帮助快速定位可能存在问题的链接,或那些需要增加实例(更长的模拟时间/更多的 Lambda 窗口)以改善预测结果、降低误差的链接。
在实际的药物发现项目中,这18个化合物将是在基准骨架基础上的新设计,探索活性位点的新区域,可能形成不同的蛋白质-配体接触。因此,在相对FEP中,我们假设尽管新设计发生了变化,但从基准到生产模式,相同的结合模式和几何结构得以保留,因此这可能解释了生产运行预测中的一些滞后性。我们要强调的是,FEP应作为更广泛的测试过程和发现工作流程的一部分使用。例如,用户可以推荐前十名结合配体以及一个来自数据集中间、预测活性较为模糊的配体。这些分子将进入检测以获得其实验活性,并可能选取一两个进行进一步的晶体学研究,以确定任何蛋白质重排,确认建模中所见的情况。即使作为更大工作流程的一部分,FEP仍然提供了对大量同类化合物进行分类的机会,识别出应进行进一步测试的子集,从而大大减少了合成弱结合配体所需的时间和资金。
此外,在Flare中,用户可以研究数据集中预测最佳和最差的结合配体进行建模,从而更好地理解这些化合物为何具有不同的活性。例如,GIST和3D-RISM\(^{6,8}\)都是Flare中的水分析工具,可以提供有关FEP计算中观察到的活性或接触的更多信息。要了解更多关于这些技术的信息,请观看我们的网络研讨会《理解水行为以增强药物设计》。以数据集中最差的结合配体Liang_15为例,我们可以将平衡后的复合物转移回主Flare项目,以了解为何它被预测为最差的结合配体,研究其结合姿态以及可能采用的不同扭转角/构象(例如在MD模拟中)。此类工作可以通过进一步理解为何该官能团未能改善活性,为下一轮设计提供信息。对于Liang_15,其活性降低很可能是因为R基团(CH₂N(CH₃)₂)无法在发生取代的水暴露区域形成相互作用。
结论
在本案例研究中,我们展示了如何利用FEP来帮助项目优先选择设计,重点关注内在活性(或结合),减少项目中弱化合物(或差结合配体)的合成数量。这种技术可以通过帮助优先选择有意义的化合物来推动项目向活性目标前进,从而加速项目进展。当然,您也可以使用它来推动那些旨在解决除了内在靶点活性之外其他问题的设计。例如,使用这种方法可以建议是否值得探索一个具有特别挑战性化学性质的化合物,即预测的活性/结合是否足够好以推进到合成阶段(如Liang_4),或者是否值得探索某些配体为何是较差的结合配体,以便更好地为未来的设计提供信息(如Liang_15)。在此,我们展示了Flare FEP在生产运行中的实际使用案例,提出了如何分析此类结果以及随后在更广泛的先导化合物优化工作流程中使用这些结果的方法。
参考文献
- Kuhn M., et al. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60, 6, 3120–3130. https://doi.org/10.1021/acs.jcim.0c00165
- Liang J., et al. Lead Optimization of a 4‑Aminopyridine Benzamide Scaffold To Identify Potent, Selective, and Orally Bioavailable TYK2 Inhibitors, J. Med. Chem. 2013, 56, 4521−4536. https://dx.doi.org/10.1021/jm400266t
- Liang J., et al. Lead identification of novel and selective TYK2 inhibitors, European Journal of Medicinal Chemistry, 2013, 67, 175 – 187. https://doi.org/10.1016/j.ejmech.2013.03.070
- Ramakrisna C., et al. Tyrosine kinase 2 inhibitors in autoimmune diseases, Autoimmunity Reviews, 2024, 23 (11), 103649, ISSN 1568-9972, https://doi.org/10.1016/j.autrev.2024.103649.
- Wang L., et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field, J. Am. Chem. Soc. 2015, 137, 2695−2703. DOI: 10.1021/ja512751q
- Luchko, T. et al.; Three-Dimensional Molecular Theory of Solvation Coupled with Molecular Dynamics in Amber, J. Chem. Theory Comput. 2010, 6 (3), 607–624
- Hahn, D. et al.; Best Practices for Constructing, Preparing, and Evaluating Protein-Ligand Binding Affinity Benchmarks [Article v1.0]. Living Journal of Computational Molecular Science. 2022, 4 (1), 1497. DOI: 10.33011/livecoms.4.1.1497
- Vinter, J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J. Computer-Aided Mol. Des. 1994, 8, 653–668. DOI: 10.1007/BF00124013.


