
摘要:键解离能(Bond Disscociation Energy, BDE)一般被定义为分子中化学键断裂过程的反应焓变,它反映了键断裂过程所需要的能量。本文介绍了BDE计算的原理,并演示了如何用Gaussian的密度泛函理论来计算化合物的键解离能。
作者:陈宇
时间:2017-11-05
一. 键解离能的计算原理
1. 基本概念
键解离能(Bond Dissociation Energy,BDE)一般被定义为分子中化学键断裂过程的反应焓变,它反映了键断裂过程所需要的能量。当分子只包含双原子时,键解离能又叫做键能。如果包含两个以上的原子,那么同种类型的化学键解离能并不一定是相等的,此时的键能称为平均键能,由分子中所有同种类型化学键解离能的平均值确定。多原子分子的键解离能包含固有键能和键断裂后分子片段的重组能两部分。
2. 计算原理
以反应1为例,分子AB可以通过均裂生成两个自由基片段A∙和B∙[1]。
|
A-B → A. + B. |
(1) |
在反应过程中,键解离能(BDE)能够由表达式2给出:
|
ΔrxnHT(1) = ΔfHT(A.) + ΔfHT(B.) – ΔfHT(AB) = DHT(AB) |
(2) |
表达式中第一项是解离过程的反应焓变,也被定义为键解离能(BDE),中间三项是三个分子片段的生成焓,反应温度为T。
一个特殊情况是当温度T = 0 K时:
由于,
|
H = U + pV |
(3) |
|
U ≅ E0 = EElec + ZPE |
(4) |
|
pV = nRT |
(5) |
因此,表达式2能够写成如下形式:
|
D0(AB) = E0(A.) + E0(B.) – E0(AB) |
(6) |
其中,方程3为焓的定义式;方程4表示温度0 K时,内能表示为基态电子能与零点振动能之和;方程5为理想状态方程;D0(AB) 为键解离能。
二. Gaussian计算键解离能的操作步骤
我们以乙烷的C-H键裂解为例(反应7),演示计算键解离能的具体步骤。
|
CH3CH3 → CH3CH2․ + H․ |
(7) |
1. CH3CH3结构优化与频率计算
首先构建初始反应物乙烷分子的结构(可以使用GaussView进行操作),然后进行结构优化,找到能量最低的构象。
输入文件如下:
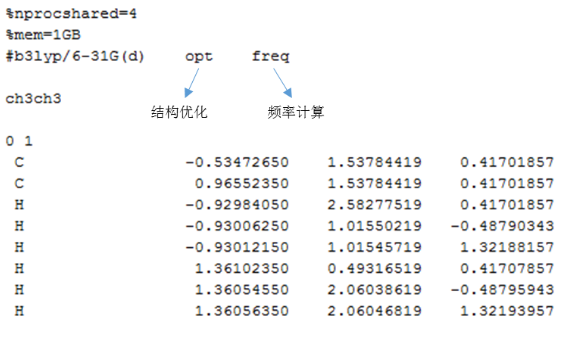
输出如下:

2. CH3CH2․结构优化与频率计算
在第一步优化的结构基础上,分别构建乙基自由基(对于较大体系这样可以减小计算量)和氢自由基片段,再对乙基自由基进行结构优化,并找到能量最低的构象(此例中氢自由基只有一个原子,所以不做结构优化,直接计算热化学性质)。
需要注意的是:由于优化的是乙基自由基,所以自旋多重度应该设定为2,并且使用非限制性波函数进行计算(当电子数为奇数时,这是默认选项)。
CH3CH2․结构优化与频率计算的输入文件如下:

乙基自由基的热化学性质输出如下:

3. H․的热化学性质计算
此例中氢自由基只有一个原子,所以不做结构优化,直接计算热化学性质。
计算氢自由基热化学性质的输入文件:
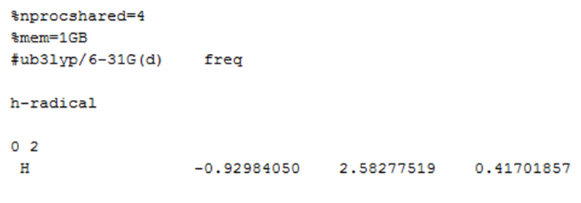
氢自由基的输出:

4. 数据分析
在Gaussian计算的输出文件中找到高亮的部分,就是分子的生成焓(需要注意的是Gaussian计算中默认的温度T = 298.15 K)。
| Items | ΔfH298 | Unit |
|---|---|---|
| CH3CH3 | -79.750759 | Hatree |
| CH3CH2․ | -79.093319 | Hatree |
| H․ | -0.497912 | Hatree |
将生成焓的数值带入公式2中可得:
ΔrxnH298(7) = ΔfH298(CH3CH2․) + ΔfH298(H․) – ΔfH298(CH3CH3) = DH298(CH3CH3)
=(-79.093319) + (-0.497912) – (-79.750759)
=0.1595 Hatree
=100.1 Kcal/mol
数据表明在所选用的方法基组下,乙烷的C-H键解离能为:
DH298(CH3CH3) = 100.1 Kcal/mol
乙烷CH3C2-H的键解离能实验值为101.1Kcal/mol[1],计算值与实验值很接近。
三. 文献
- Blanksby, S. J.; Ellison, G. B. Bond Dissociation Energies of Organic Molecules. Accounts of Chemical Research 2003, 36, 255-263. DOI:10.1021/ar020230d
- Bach, R. D.; Dmitrenko, O. Strain Energy of Small Ring Hydrocarbons. Influence of C−H Bond Dissociation Energies. Journal of the American Chemical Society 2004, 126(13): 4444-4452. DOI:10.1021/ja036309a
四. 相关主题
Gaussian教程|pKa的计算:http://blog.molcalx.com.cn/2017/10/15/gaussian-pka-calculation-tutorial.html
五. 联系我们
广州市墨灵格信息科技有限公司是Gaussian的代理。当前最新版本为Gaussian 16 A.03, 如果需要采购软件,请联系我们。


