
摘要:Gaussian ONIOM可以用来模拟大分子体系,可以是有机化合物或生物大分子。本教程介绍了ONIOM计算的基本思想,并通过丙醛分子的基态结构详细说明了如何利用GaussView进行分子的分层、计算路径的设定、并利用Gaussian程序进行有机化合物的ONIOM计算。
作者:陈宇
日期:2018-01-15
一.ONIOM方法的基本原理
1. 基本思想
在实际的研究中,真实的分子体系可能包含大量的原子,对于这种体系而言,由于计算资源和计算时间的限制,若使用较高精度的模型方法进行模拟往往是不实际的,ONIOM方法的思想在于把一个分子体系分为若干个区域(也称为层,通常可以分成2层和3层),不同的层能够使用不同精度的方法进行计算,如图1所示。
若划分为2层:则通常划分为High Layer(高层)和Low Layer(低层)。其中高层包含感兴趣的研究区域(对于化学反应而言,则是断键和成键区域),这部分需要使用最昂贵的计算方法;而低层则是其余的部分,这部分则使用便宜的计算方法。
3层划分:有些情况下,反应中心受环境的影响较大,此时低水平方法不能有效的描述这些效应,因此还需要增加一个Medium Layer(中间层,这一层通常紧邻高层系统),这一部分采用介于高层和低层之间的方法进行描述。
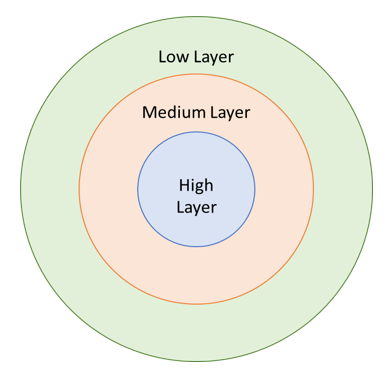
图1. ONIOM分层原理
2. ONIOM计算电子能的原理
由于在ONIOM计算中,不同的层使用不同精度的方法进行计算,因此相比于传统计算,对于体系的电子能,ONIOM有更复杂的计算步骤,图2中每一个圆表示相应的方法在相应的层系统下计算得到的电子能,加号表示在ONIOM能量表达式中加上这个能量项,减号则表示减去这个能量项。
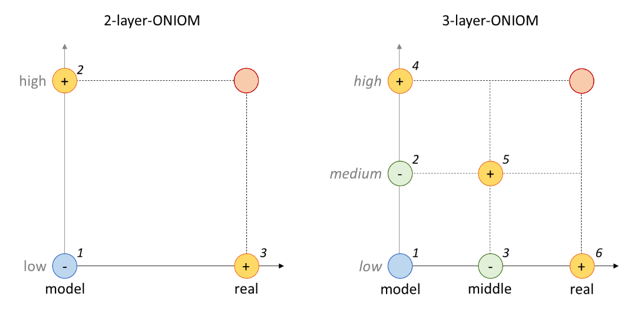
图2. 电子能计算原理
对于2-layer体系:
E(ONIOM) = E(Low, Real) + E(High, Model) – E(Low, Model)
对于3-layer体系:
E(ONIOM) = E(Low, Real) + E(Medium, Middle) + E(High, Model) – E(Medium, Model) – E(Low, Middle)
为了进行一个ONIOM计算需要进行如下三个步骤:
- 利用GaussView对分子体系进行分层
- 构建ONIOM的输入文件
- 结果分析
二. 利用Gaussian程序进行ONIOM计算分层方案
在这个教程中,我们以丙醛分子(图3)的结构优化为例,说明如何进行ONIOM的计算。我们只演示2层的ONIOM计算(3层计算的步骤完全相同,只是需要再划分一个中间层,而划分方法与划分低层的方法相同)。
图3. 丙醛分子
通常需要注意的是:为了使ONIOM计算更好的模拟真实体系,在对分子体系进行分层时,需要考虑一下几个原则:
- 截断的化学键应该是单键(同时应该是非极性键),而不能是双键或三键
- 截断的化学键不能是成环的化学键
- 分子中有共轭作用的部分要划分到同一层
- 在化学反应中,涉及断键和成键的部分要划分到同一层
丙醛分子的2层ONIOM计算可以按图4的分层方案来实现:
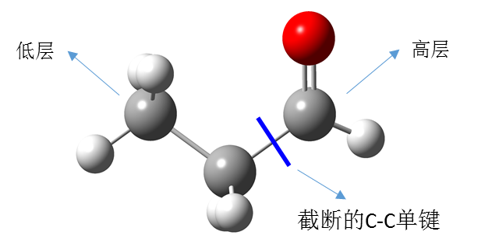
图4. 分子层的划分
三. 丙醛2层ONIOM计算的详细操作步骤
利用GaussView对分子体系进行分层,下面我们将详细说明这个过程:
1. 构建丙醛分子
首先在GaussView中建立丙醛分子模型。
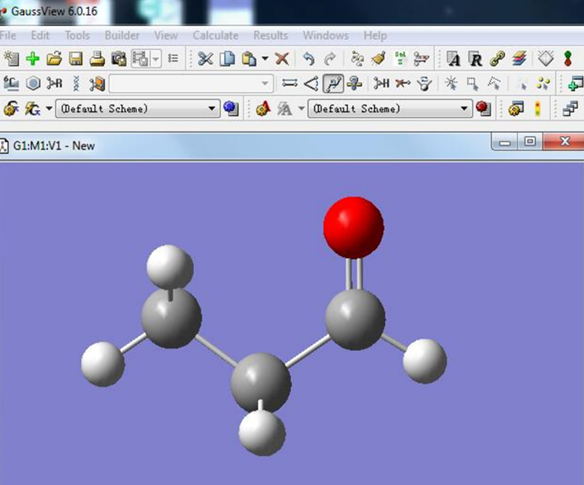
图5. 丙醛分子的模型
2.用GaussView的Atom Group Editor进行ONIOM Layer编辑
在菜单栏中Tools的下拉菜单中点击Atom Groups调出原子团编辑对话框,然后在原子团(Atom Group)类型下拉菜单中选择ONIOM layer选项。此时可以看出所有的原子都处于高层,如图6所示。
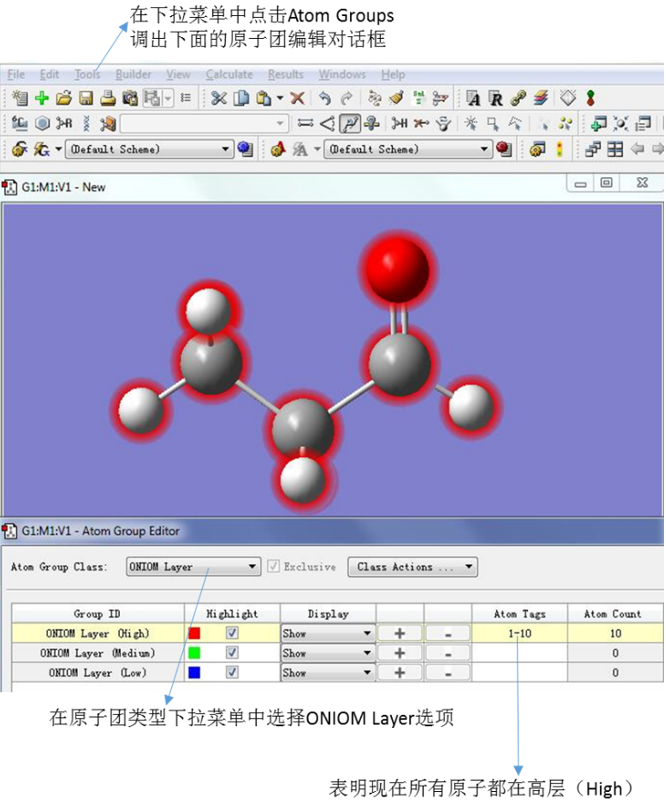
图6. ONIOM Layer的编辑
3. 设置低层原子
如图7,为了设置低层部分,点击圈选工具,选中处于想要设置为低层(Low)的原子(也就是1-7号原子,原子标号可以通过点击View下拉菜单中Labels选项显示),然后点击“ONIOM layer (Low)”所在行的“+”号,此时显示1-7号原子处于低层,即完成我们想要的分层。
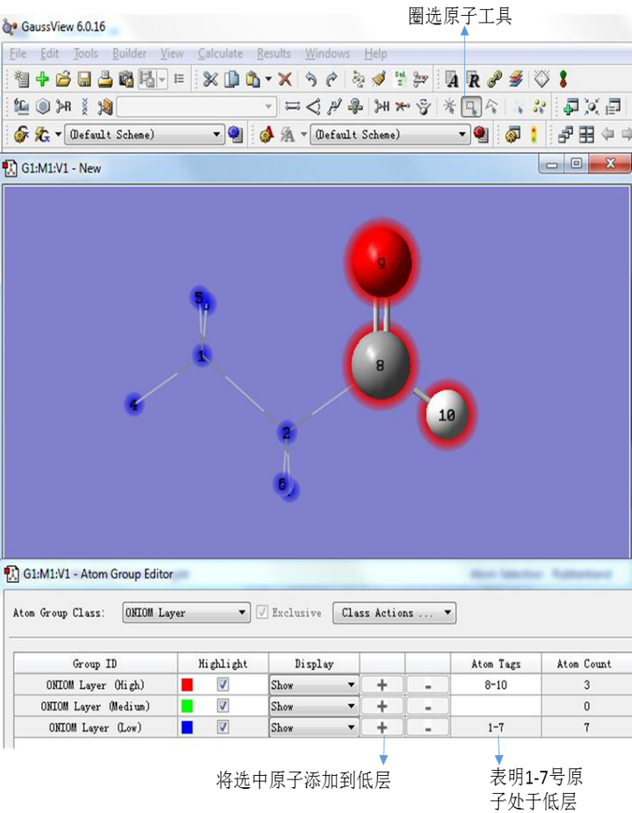
图7. 设置ONIOM低层原子
4. 完成ONIOM分层
点击原子团编辑对话框中下面的“OK”关闭对话框,然后点击工具栏中Calculate下拉菜单中的Gaussian Calculation Setup选项,调出ONIOM计算路径设置对话框。
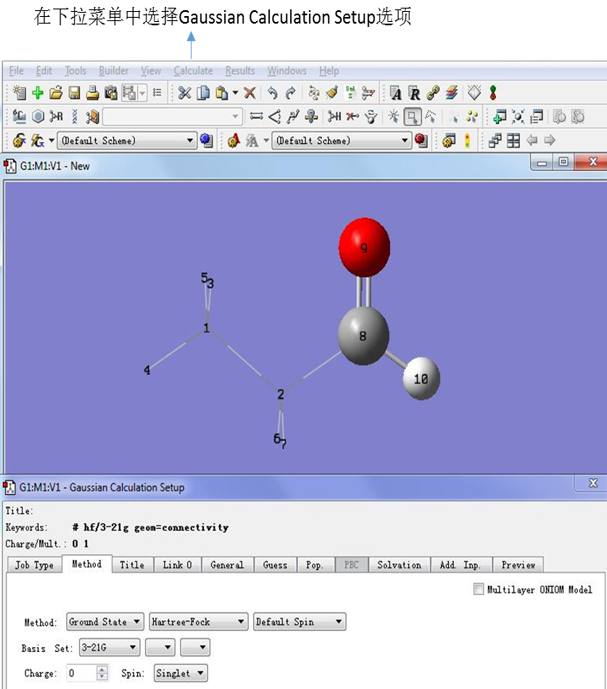
图8. 完成ONIOM分层返回GaussView的计算界面
5. 设定作业类型为:优化与频率计算
点击Job Type,进入工作类型设置界面,选中opt+freq,进行结构优化和频率计算,选择minimum以优化极小点。
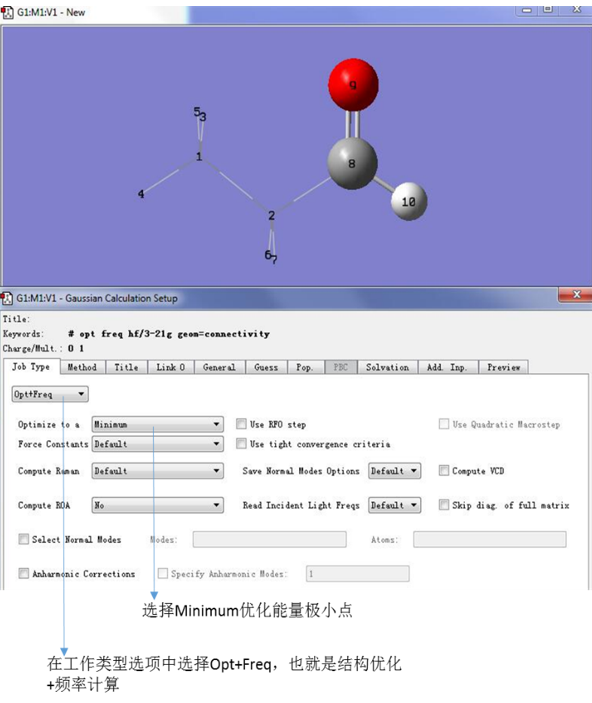
图9. 设定计算类型为Opt + Freq
6. 设定ONIOM High Layer的计算方法
点击Method,进入方法基组设定界面,然后勾选右上侧的Mutilayer ONIOM Model,进入ONIOM计算路径编辑界面。首先,点击High Layer,对高层部分进行方法基组的设定(在这个教程中,我们为高层部分指定图10中的方法基组)。可以看到,设定的方法基组已经被显示在对话框的上方。
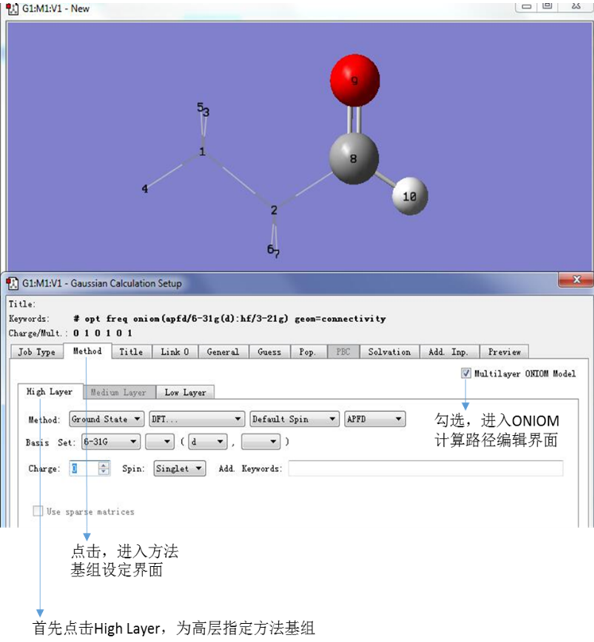
图10. 设定High Layer的计算方法
7. 设定Low Layer的计算方法
点击Low Layer,为低层指定方法基组,我们为低层部分指定图11中的方法基组。
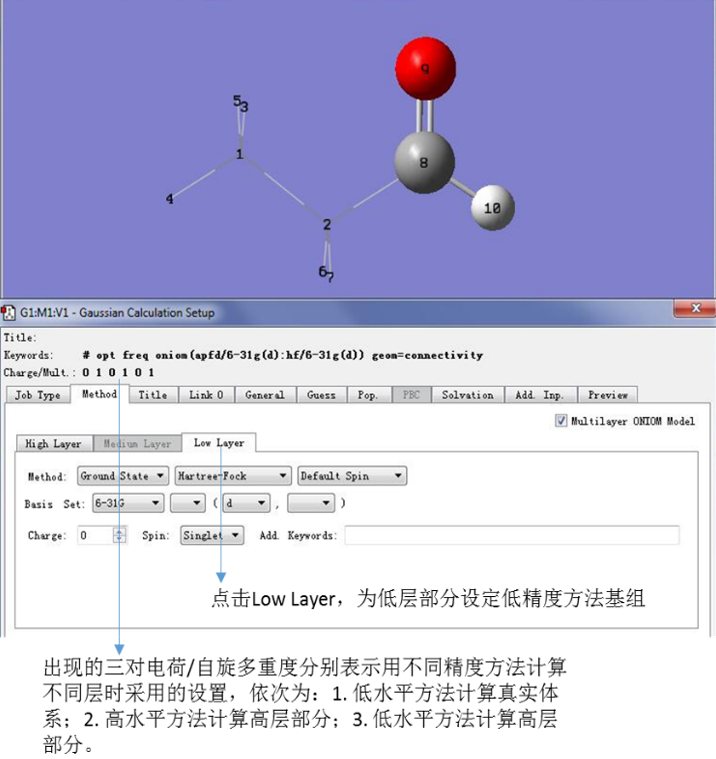
图11. 设定Low Layer的计算方法
在这里,我们注意到出现了三对电荷/自旋多重度,它们分别表示用不同精度方法计算不同层时采用的设置,依次为:1. 低水平方法计算真实体系;2. 高水平方法计算高层部分;3. 低水平方法计算高层部分。
8.生成Gaussian输入文件,并编辑
点击对话框下面的Edit,保存文件,然后弹出文本编辑界面。如果有必要,可以直接在其中编辑输入文件。在这个教程里,我们将最终的输入文件改写成如下形式:
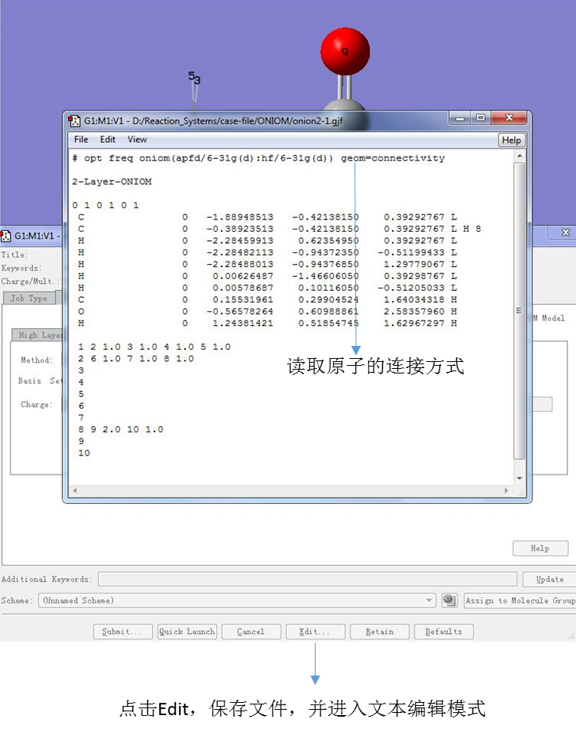
图12. 编辑输入文件
9.保存输入文件
保存更改的输入文件,即可完成输入文件的编辑。
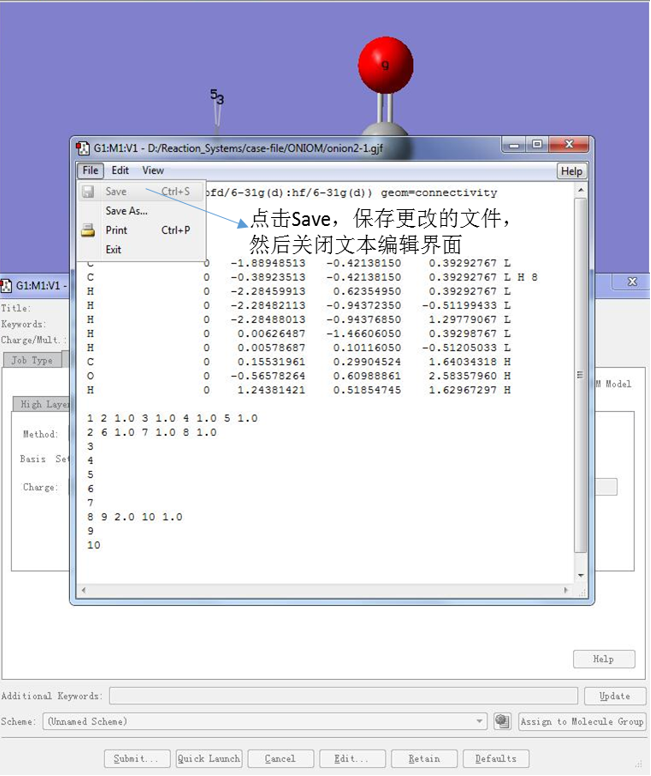
图13 保存输入文件
10. 提交输入文件,进行计算
11. 结果分析
(1)频率分析
与通常的Gaussian极小点结构优化一样,振动频率中应该没有虚频。
(2)ONIOM电子能量
图14给出了输出文件中电子能量的结果,根据上面的公式,我们能够得到推测的ONIOM电子能:
ONIOM (extrapolated energy) = -192.489635432370
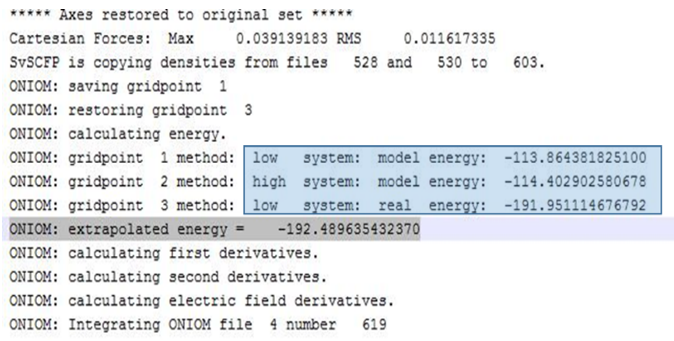
图14. ONIOM计算的电子能量
四. 参考材料
- http://gaussian.com/oniom
- James B. Foresman and Aeleen Frisch. Exploring Chemistry with Electronic Structure Methods. Third Edition. P61-64
- http://gaussian.com/oniom_technote
- 小分子与生物大分子相互作用的OMIOM(QM/MM)模拟
Expl Chem3第61-64页提供了ONIOM计算有机化合物的详细过程
本文详细地介绍了Gaussian的ONIOM技术。
请参阅James B. Foresman and Aeleen Frisch. Exploring Chemistry with Electronic Structure Methods. Third Edition.第401-461页教程


