
摘要:大环化(Macrocyclization)在制药工业药物发现领域起着至关重要的作用,分子大环化可以帮助您:1)获取更佳的类药性质;2)改善靶标选择性;3)增强结合亲和力。本文与大家分享一个用SPARK分子大环化策略来设计BRD4抑制剂的案例。在本案例中,SPARK可以成功地设计出与报道的BRD4大环抑制剂相同或非常相似的大环化合物,SPARK生成的连接臂大小的分布与实验SAR数据非常吻合。SPARK的分子大环化向导是一种快速且易于使用的工作流程,它可生成有意义且多样化的计算结果,可以用于指导大环药物的发现。
原文: Matthias Baue,2018-02-22,《用SPARK设计大环BRD4抑制剂》
编译:肖高铿
一. 大环化(Macrocyclization)
分子的大环化有诸多优势,比如提供多样的官能团、改善类药性(尤其是多肽药物的大环化拟似物)、增加选择性2。分子大环化可以将分子锁定于一个低能结合构象、并减少结合熵变损失,因此可以用于提高化合物的结合亲和力2–4。分子大环化的难点在于分子的合成可行性,因此在合成前预测分子大环化策略的有效性是大环药物发现成功的关键。
最近,Wang等人报道了一类新的基于吡啶酮的BRD4 Bromodomain抑制剂,在细胞系和异种移植模型中显示出有希望的抗癌作用4 。从吡啶酮片段苗头化合物开始,采用基于结构的设计进一步优化化合物,得到活性为个位数纳摩尔水平的化合物;分子大环化进一步改善了化合物的结合亲和力、细胞水平活性和药代动力学性质(Figure 1)4。
在这个案例中,我们使用SPARK–Cresset的生物电子等排体和估计跃迁工具来设计非大环吡啶酮BRD4抑制剂的大环化策略,并根据Wang等人报道的实验数据来评估设计结果。
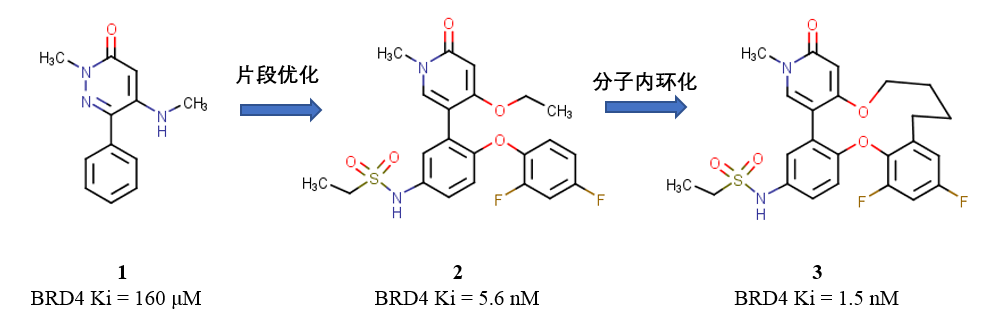
图1. Wang等人报道的基于吡啶的BRD4 Bromodomain抑制剂开发。 将优化过的片段[2]环化,结果显示结合亲和力提高了约4倍(用TR-FRET结合分析法测定的Ki值)。 除了结合亲和力得以提高之外,化合物[3]还提高了细胞水平活性并改善了药代动力学性质。
二. 分子大环化的方法
SPARK利用具有相似静电和空间特性的片段来替换给定起始分子中的特定部分5, 其中静电和空间的相似性比较是基于XED力场计算得到的分子场来计算的6-8。SPARK的一个主要好处是它在搜索替换片段时保留了大量的3D信息:考虑了整体分子的电子和构象效应,不是仅仅对相应的替换片段进行评分,而是对整个最终分子进行评分。替换片段是从SPARK的片段数据库中筛选出来的,这些数据库又是从可供商品销售的化合物数据库或文献报道的化合物生成的。当一个片段被识别为含有所需数量的连接点并具有与起始分子相似的形状/几何形状的片段之后,SPARK会使用XED力场对分子进行能量最小化计算以去除不良的碰撞和不利的几何形状,然后再进行最终分子的场计算和相似性优化以得出最终的打分值。
BRD4大环抑制剂的设计采用SPARK的分子大环化向导来实现。向导模块提供了快速且易于使用的图形界面工作流,包含了专为分子大环化设计而量身定制的专用参数设置(图2)。 在本实验中,我们使用了BRD4与非大环化合物[2](PDB 5UEY)的复合物晶体结构,并以复合物中的化合物[2]作为起始分子(图2)。 [2]的生物活性构象表明,吡啶酮骨架的乙氧基基团靠近2,4-二氟苯氧基环(图3)。 另外,在结合位点里的配体部分地暴露于溶剂,可以提供足够的空间以容纳额外的连接基团原子。 因此,我们选择了连接点1(乙氧基片段)和在2,4-二氟苯氧基环的6位的连接点2(见图3)。
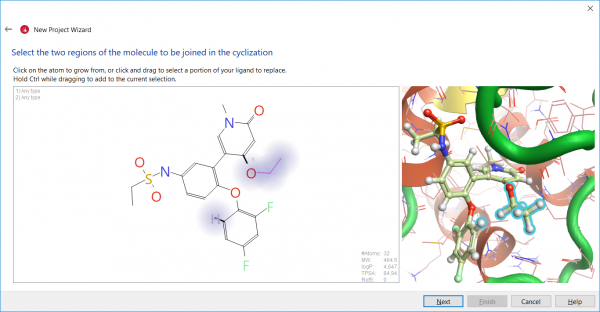
图2. 在分子大环化向导里选择连接点。连接点1是乙氧基,连接点2是2,4-二氟苯氧环的6位氢原子。右边的3D视窗可以实时观察连接点以确保连接点的选择是正确的。
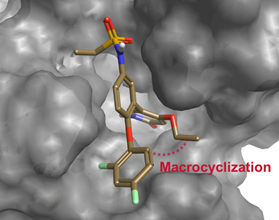
图3. 抑制剂[2]与BRD4的复合物结构(PDB 5UEY)。乙氧基与2,5-二氟苯氧基在空间上很靠近并且有部分暴露在溶剂中,这意味着我们的连接点选择是可行的。
为了让连接臂的设计偏向于已有的化合物,将氧指定为连接点1的连接原子,第二个连接点则不受约束。 此外,还将受体结构设置为排除体积来指导连接臂设计,并且还对与受体形成氢键相互作用的羰基和磺酰基的场点加上约束。该实验使用专用的“Ligand Joining / Macrocyclization”设置参数对片段数据库CHEMBL_common,Common和VeryCommon(其中共包含大约120K个片段)进行搜索。 最后将SPARK计算获得的结果导入到Flare9中进行可视化分析。
三. 结果与讨论
本研究的分子大环化计算产生了500个大环化合物。大环化合物[3]与受体的复合物结构(图4)表明:环化后的分子依然保留了与BRD4口袋的关键相互作用,然而,比之化合物[2],化合物[3]的构象略微不同, 特别是2,4-二氟苯氧基环部分。 尽管存在这种构象变化,最高得分的SPARK结果包含了几个与化合物[3]结构非常相似大环化合物(见图5),例如具有丁醇(排序11),丙-1,3-二醇(排序9),4-氨基丁醇 -1-醇(排序2),丁-2-醇(排序4)或3-丁烯-1-醇连接臂(排序17)。 化合物[3]也被SPARK设计出来,排序59(见图5)。
在SPARK打分最高结果中富集了很多具有4-6个原子的连接臂(表1)的大环化合物。 这一发现与Wang等人报道的实验数据非常吻合。他们发现,与5个原子的连接臂相比,仅有3个原子的、短的大环连接臂的化合物其结合亲和力比5原子连接臂化合物降低了大约50倍。
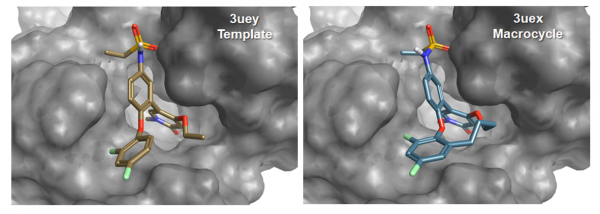
图4. BRD4与化合物[2](PDB 5UEY)以及化合物[3](PDB 5UEX)的复合物结构
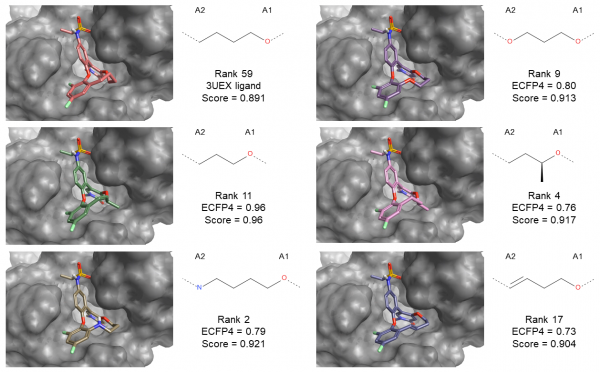
图5. SPARK分子内环化实验结果。在打分最高的结果中,SPARK设计出了几种连接臂要么与化合物[3]一样,要么非常近似。ECFP4值是与化合物[3]的Tanimoto系数。SPARK的打分值用于给化合物排序。连接点A1、A2见方法部分的说明。
比之五原子连接臂,四原子连接臂显示出轻微的亲和力降低,而六原子的连接臂结合亲和力与之相当4。
图6给出了打分最佳结果的连接臂大小。有趣的是,对于较短的连接臂(例如,具有4个连接原子的丁-2-醇),SPARK预测的片段模拟了乙氧基侧链的疏水体积[2]。 对于三原子连接臂的大环化合物,实验结合亲和力的降低可能是因为这部分疏水的贡献被遗漏了。 因此,添加一个甲基到化合物2的接头可以提高短臂大环化合物对BRD4结合亲和力(例如图6中排序为6的化合物)。
表1: 打分最高的前10、25与50的连接臂大小分布
| Top N results | 3 linker atoms | 4 linker atoms | 5 linker atoms | 6 linker atoms | 7 linker atoms |
| 10 | 2 | 6 | 1 | 1 | 0 |
| 25 | 2 | 16 | 4 | 8 | 0 |
| 50 | 2 | 32 | 12 | 9 | 0 |
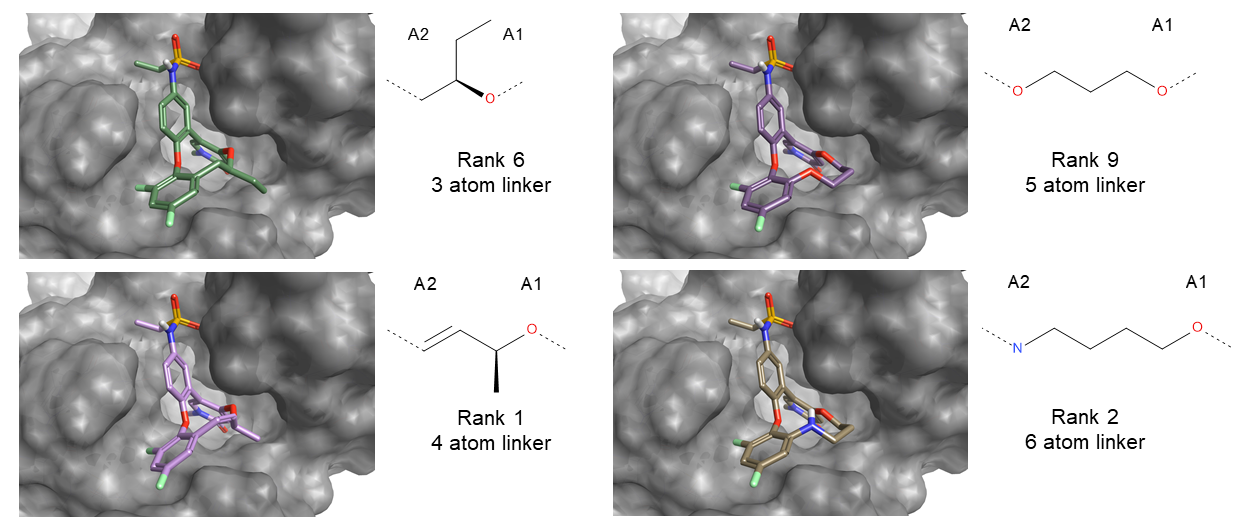
图6. SPARK分子大环化计算结果。在打分最高的10个结果中,SPARK设计出的连接臂大小为3-6个原子。分别给出了每个不同大小的连接臂化合物中打分最佳的结果。连接点A1与A2详见方法部分。
四. 结论
本案例研究表明,SPARK可以成功地设计出与报道的BRD4大环抑制剂相同或非常相似的大环化合物4 。SPARK生成的连接臂大小的分布与实验SAR数据非常吻合4。
SPARK的分子大环化向导是一种快速且易于使用的工作流程,它可生成有意义且多样化的计算结果,可以用于指导大环药物的发现。
五. 参考文献
- http://www.cresset-group.com/spark
- Wagner, V.; Rarey, M; Christ, C.D. Computational Macrocyclization: From de Novo Macrocycle Generation to Binding Affinity Estimation. ChemMedChem 2017, 12 (22), 1866–1872.
- Yu, X.; Sun, D. Macrocyclic Drugs and Synthetic Methodologies toward Macrocycles. Molecules 2013.
- Wang, L.; McDaniel, K. F.; Kati, W. M. Fragment-Based, Structure-Enabled Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extra-Terminal Domain (BET) Family Bromodomain Inhibitors. J. Med. Chem. 2017, 60 (9), 3828–3850.
- Tosco, P.; Mackey, M. Lessons and Successes in the Use of Molecular Fields. In Comprehensive Medicinal Chemistry III; Elsevier, 2017; pp 253–296.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. J Comput Aided Mol Des 1994, 8 (6), 653–668.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. Chem. Inf. Model. 2006, 46 (2), 665–676.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. II. Organic Complexes. J Comput Aided Mol Des 1996, 10 (5), 417–426.
- http://www.cresset-group.com/flare
六. 接下来可以做什么
- 进行结合亲和力预测、排序以评估哪个化合物最值的合成
- 评估大环化合物的结合构象稳定性,以决定是否值得进一步合成该大环化合物。
- 用3D-QSAR预测化合物活性
将计算结果发送给FLARE,采用WaterSwap的FEP,TI等方法预测结合亲和力,参见:《FLARE教程 | WaterSwap计算结合自由能》《FLARE案例 | WaterSwap计算BRD4抑制剂的结合自由能》
即使计算比较昂贵FEP等方法不能完全采集化合物的全部构象,振动模式也未被考虑进去,因此有必要进一步评估化合物结构构象的稳定性,具体方法参见:《构象稳定性评估及其在先导化合物优化中的应用》
将计算结果发送给FORGE,用3D-QSAR模型预测化合物活性。参见:《Forge教程 | 3D-QSAR建模》


