
摘要:本文介绍了关于过渡金属催化的相关概念,并以金属钯和醋酸根配体形成的催化剂为例,阐述了过渡金属催化机理研究中需要考虑的关键问题,并详细介绍了芳基Csp2-H键的几种断裂模式,并演示了如何用Gaussian软件对这些模式进行了过渡态计算。
–钯(醋酸根作为配体)催化的芳基Csp2-H键活化
作者:陈宇
日期:2019-01-17
一. 前言
对化学反应的研究是化学研究领域最核心的内容,不仅在工业,生物,医药等领域能看到化学反应的身影,甚至整个宇宙空间可以看作是一个巨大的反应容器,其中不断地进行各种化学反应过程。
化学反应过程是化学键断裂/生成的过程,从表观来看,就是一种或几种分子在一定条件下转变成另外一些分子的过程。在早期的化学研究中,由于研究工具的限制,只能通过对表观物质转变的观察总结一些经验公式,而不能对反应过程的微观状态提出更细致的阐述,其中最重要的就是包含化学键断裂和生成信息的微观状态,这些微观状态就构成了我们所谓的反应机理(也称为反应路径)。
量子力学的发展极大地推动了化学研究,基于量子力学和经典力学的计算化学的兴起便是其中之一,通过计算化学工具,能够容易的阐述反应过程的微观机制,这也使我们对化学的认识达到了一个前所未有的高度。
然而需要明白的是理解反应机理只是计算的开始,有效的设计调控才是计算的最终目的,尽管在现阶段,计算化学主要的目的仍然是辅助理解实验现象,但我们相信随着计算方法和工具的突破,计算化学作为更独立的探索新现象和未知领域的时代终将到来。而事实上,计算化学工具已经在探索未知的领域上暂露头角,比如新型材料,催化剂的设计等等。
当我们考察一个反应能否发生时,通常会考虑两方面因素:热力学和动力学。热力学决定了在特定方向上反应是否具有自发性(吉布斯自由能小于零,反应具有自发性);动力学则决定了在一定条件下,反应速度的快慢。也就是说即便反应在热力学上具有自发性,但如果反应在动力学上速度很慢,也无法在特定条件下获得目标产物,氢气和氧气反应生成水的过程就是一例,这个过程是自发性过程,但速度很慢,因此在室温条件下,我们不能观察到生成水的现象。
对于上述动力学问题,一个重要的解决方案就是引入催化剂(图1),催化剂的引入不会改变反应的热力学,却能改变反应的速度(也就是动力学)。从微观反应机理的角度理解,催化剂实际上影响了化学键的断裂和生成过程,使得反应经历了能量更低的过渡态。
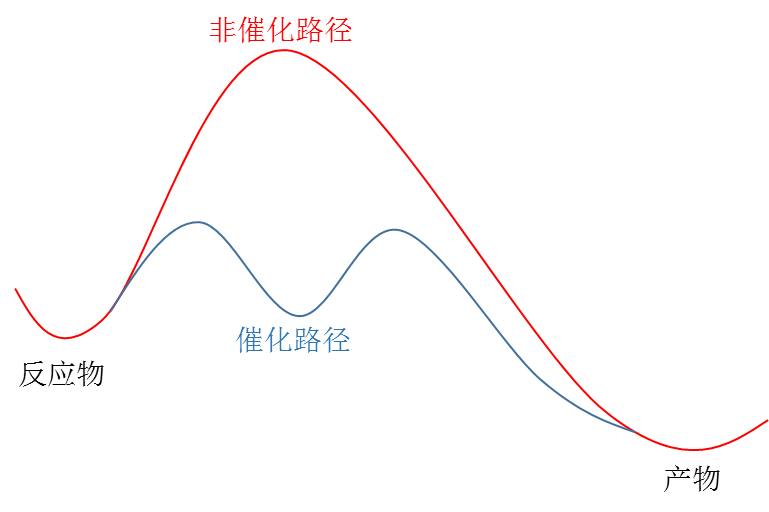
图1. 催化剂使反应经历了能量更低的过渡态
基于上面的阐述,我们很容易看出计算化学工具在催化剂的设计和调控上有巨大的用武之地,通过对反应过程微观状态的详细描述,我们能够深入理解催化剂在反应过程中如何影响化学键的断裂和生成,利用这些信息,可以通过调整催化剂的结构,使生成目标产物的反应路径更加有利。
过渡金属配合物是最重要的一类催化剂,已经被用于各种各样的反应类型,在这个系列的教程中,我们简要介绍不同类型过渡金属配合物催化的不同反应类型中涉及的关键步骤。
如图2所示,过渡金属催化系统包含了三个重要组成部分:金属类型,配体类型和添加物。其中金属和配体决定了催化剂的结构(不同的催化剂结构会导致不同的断键/成键方式),而添加物通常参与到催化剂重新生成的过程中(有时也会参与到反应过程中)。概括来说,催化剂能够影响反应的化学选择性,区域选择性和立体选择性,这也是设计催化剂时需要考虑的关键问题。
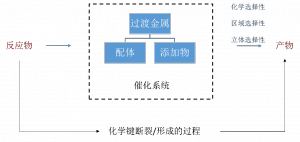
图2. 过渡金属催化系统包含了三个重要组成部分
催化剂结构能够显著影响催化反应的机理,因此对催化剂结构的了解是探究反应机理重要的环节之一。对于某一种类型的过渡金属而言,其形成的金属配合物(在这里指催化剂)的结构完全依赖于配体的类型,简而言之,就是不同(一个催化系统中,配体的种类可能不止一种)类型的配体与某一类型的过渡金属在特定的条件下能够形成具有特定结构的金属配合物,当这种金属配合物能够改变某个反应的动力学过程时,它就成了这种反应的催化剂。
在这个系列的教程中,我们每篇教程都遵照如下的模式:选用一种过渡金属,一组配体和一种反应类型,然后详细讨论由过渡金属和配体形成的金属配合物是如何影响这种反应的断键/成键过程的。
在研究催化反应机理时,通常会遵循如下顺序:先确定催化剂的结构,再确定断键和成键模式。催化剂结构的对错直接决定是否能够获得恰当的反应机理。
在这篇教程中,我们选用金属钯(Pd)和醋酸根(作为配体)形成的金属配合物作为催化剂,讨论它如何影响芳香烃的Csp2-H键的断裂(图3)。
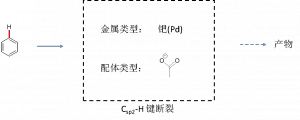
图3 芳香烃的Csp2-H键的断裂
二.确定金属配合物的结构
确定金属配合物的结构是研究反应机理至关重要的一步,它直接决定了以此为基础得到的反应机理是不是合理。寻找金属配合物存在形式的基本原则是:能量最低原则。通常配体与金属会有多种配位形式,但在平衡状态下,根据玻尔兹曼分布,能量最低的配合物会占据大多数,但需要记住的是,能量最低的配合物不一定就是催化剂,它需要被激活,才能完成催化过程,通常激活的过程就是配合物中某个配位点解离的过程。
过渡金属呈现的价态对判断配合物结构有很大的帮助,金属钯(Pd)通常呈现的价态为零价,二价和四价,在只有醋酸根作为配体的情况下,钯通常表现出正二价,从表观上看,也就是两个醋酸根配位到二价钯中心上(整体呈中性),配合物结构为如下的形式(图4,不考虑聚合物)。
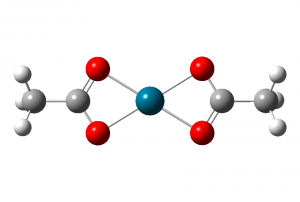
图4.两个醋酸根配位到二价钯中心上的结构
三.Csp2-H键断裂的模式
C-H键的断裂模式依赖配合物的结构,在这个体系中,我们主要介绍三种C-H键断裂模式(图5):协同金属化-去质子化机理;外源碱辅助的去质子化机理;σ-复分解机理。
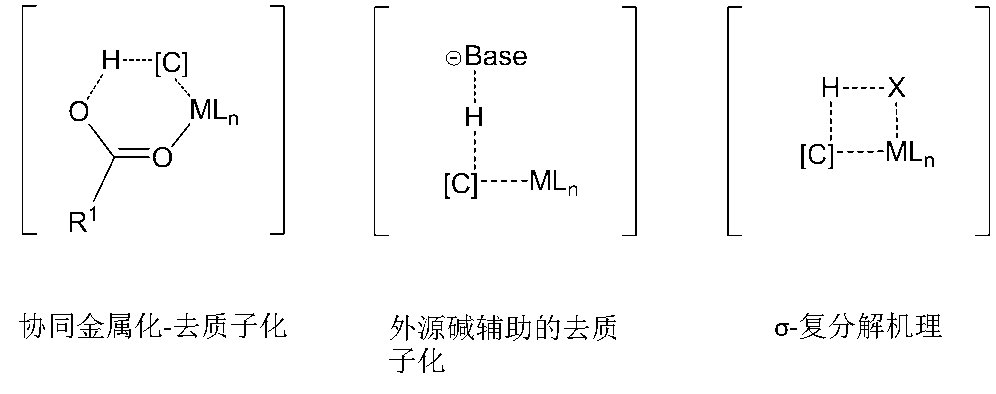
图5. 三种C-H键活化模式
接下来,我们按照图5给出的C-H键活化模式分别优化三种断裂模式下的过渡态,在这里我们只给出过渡态优化的输入文件,但为了计算反应能垒,想要练习的读者还需要优化反应物的结构,方法基组要与优化过渡态一致。寻找过渡态的具体方法参见《搜索过渡态》教程,这里不做详细论述。
3.1 协同金属化-去质子化
通常在发生协同金属化-去质子化机理的催化体系中,会存在至少有两个配位点的配体,其中一个配位点与金属配位,而另一个协助拔氢。在我们的体系中,其可能的过渡态结构如图6所示。
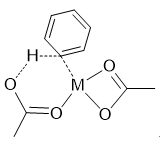
图6. 协同金属化-去质子化过渡态结构
协同金属化-去质子化过渡态优化的输入文件(见教程附件TS-CMD.gjf):
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 | %nprocshared=8 %mem=1GB # opt=(calcfc,ts,noeigen) freq b3lyp/genecp TS-CMD 0 1 Pd 0.59565400 0.36963100 0.05863000 O 1.86198100 -1.26662300 -0.17451200 C 2.90100500 -0.53519300 0.01681300 O 2.73481600 0.70458700 0.23791700 O -0.21283400 2.22711800 0.35228700 C -1.34622500 2.53909800 -0.13539300 O -2.13834200 1.72377900 -0.69333500 C 4.27120600 -1.14825500 -0.04614900 H 4.27208000 -2.11232600 0.47007400 H 4.53853300 -1.32973500 -1.09371500 H 5.00811800 -0.47546800 0.39733900 C -1.75339000 3.99454000 -0.05260300 H -1.16084400 4.51867300 0.69865400 H -1.58044300 4.46093100 -1.02920000 H -2.82006300 4.07027900 0.17130300 C -1.23988000 -0.69067200 -0.13515300 C -1.80423300 -1.07198500 1.10540700 C -2.38787900 -2.32560700 1.27472700 C -2.42315700 -3.22234500 0.20238800 C -1.88208900 -2.86719700 -1.03850300 C -1.28947000 -1.61861400 -1.20227100 H -1.61608700 0.51666400 -0.48859700 H -1.79661000 -0.36620600 1.93261500 H -2.81658100 -2.60577400 2.23320800 H -2.88024400 -4.20024600 0.33140200 H -1.92170500 -3.56707000 -1.86885700 H -0.86654800 -1.34623200 -2.16566700 C H O 0 6-31G* **** Pd 0 LanL2DZ **** Pd 0 LanL2DZ !所有空白行都是特意的,不要忽略 !注意:第3行的genecp表示这里使用赝势基组 |
图8为优化得到的过渡态结构:
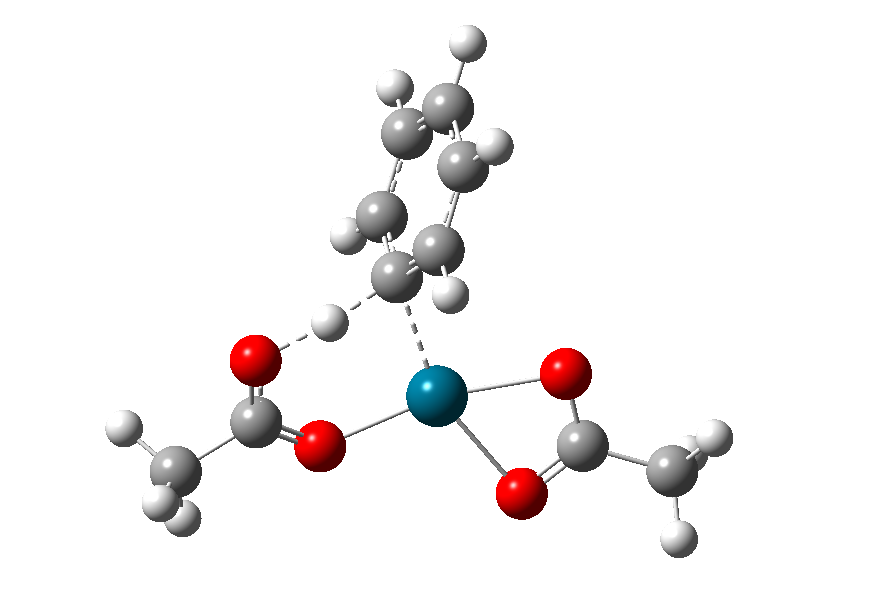
图8. 协同金属化-去质子化过渡态优化得到的结构
需要注意的是:通常计算过渡金属时需要使用赝势,具体方法参见教程《使用基组和赝势》。
3.2外源碱辅助的去质子化
在发生外源碱辅助的去质子化机理的催化体系中,则需要体系中存在布朗特斯碱(为了接受质子),在辅助劈裂C-H的过程中,外源碱不配位到过渡金属上,只有一个质子受体位点协助拔氢,在我们的体系中,其可能的过渡态结构如图9所示。
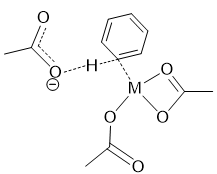
图9. 外源碱辅助去质子化可能的过渡态结构
外源碱辅助的去质子化过渡态优化的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 | %nprocshared=8 %mem=1GB # opt=(noeigen,calcfc,ts) b3lyp/genecp freq TS-Outbase -1 1 Pd 0.49923800 0.09377200 0.34821800 O -1.13652000 -0.89383300 1.21547200 C -1.93226300 0.09732200 1.04424900 O -1.51706700 1.17185200 0.52930000 O 2.24376500 0.77849000 -2.36818100 C 2.26075600 1.63445800 -1.48667800 O 1.78051700 1.53740500 -0.27893200 C -3.38182900 -0.07172600 1.44355400 H -3.86248500 0.90361700 1.55210200 H -3.90352900 -0.63968500 0.66418700 H -3.45445300 -0.64126500 2.37469400 C 2.91250300 3.00375400 -1.72574200 H 2.16133600 3.79563500 -1.62725100 H 3.68441800 3.19356100 -0.97162100 H 3.35316700 3.03767200 -2.72521200 C 1.97054400 -1.28904300 0.65013600 C 3.25062400 -1.15839000 0.06703000 C 4.36845300 -1.71154800 0.68953100 C 4.23193900 -2.39235300 1.90362400 C 2.96905900 -2.54784800 2.48310600 C 1.84501300 -2.01222400 1.85743300 H 1.04455600 -1.82565900 -0.34303400 H 3.33953400 -0.64992000 -0.88673600 H 5.34908700 -1.61469000 0.22775200 H 5.10826400 -2.81772000 2.38960400 H 2.86024200 -3.10225100 3.41270400 H 0.85574200 -2.17597900 2.27046700 C -0.11008200 -3.81536100 -0.81474800 O 0.08220200 -4.19456700 0.34233100 O 0.29730300 -2.71757500 -1.37383400 C -0.93381100 -4.68544100 -1.78451200 H -1.88616800 -4.18837100 -2.00628900 H -0.40479600 -4.80858300 -2.73605600 H -1.13370600 -5.66332700 -1.33735400 C H O 0 6-31G* **** Pd 0 LanL2DZ **** Pd 0 LanL2DZ !所有空白行都是特意的,不要忽略 !注意:第3行的genecp表示这里使用赝势基组 |
图10. 外源碱辅助的去质子化过渡态优化的输入文件
图11为优化得到的过渡态结构:
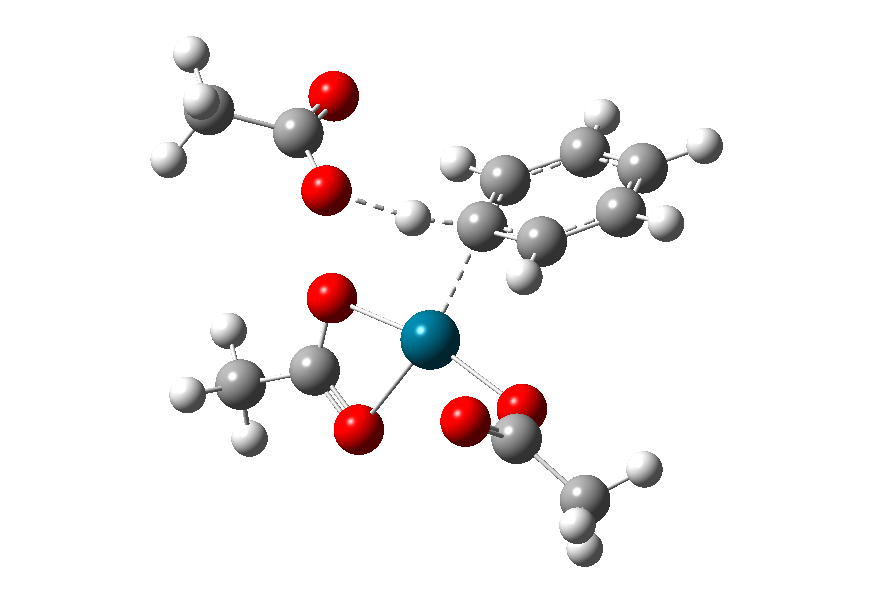
图11. 优化得到的过渡态结构
3.3.σ-复分解机理
在σ-复分解机理中,典型的形成一个四元环过渡态,其中氢直接进攻配体中与金属成键的原子,在我们的体系中,其可能的过渡态结构如图12所示:
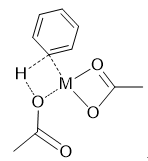
图12. 可能的过渡态结构
σ-复分解过渡态优化的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 | %nprocshared=8 %mem=1GB # opt=(noeigen,calcfc,ts) b3lyp/genecp freq TS-Metathesis 0 1 Pd 0.31866200 -0.36420400 -0.37886000 O -1.63503900 -1.10718100 -0.18306700 C -2.12225500 0.07685700 -0.23728600 O -1.32635600 1.06050400 -0.36910800 O 2.32297100 0.97180300 1.50006600 C 2.48902200 1.22045300 0.32478600 O 1.86741700 0.54521800 -0.68328400 C -3.61009100 0.28791100 -0.17731400 H -3.83518100 1.23475000 0.31980000 H -4.00716600 0.33697300 -1.19826400 H -4.09167400 -0.54370400 0.34211900 C 3.40394900 2.29641100 -0.21602900 H 4.08703300 1.87894900 -0.96246900 H 2.80486600 3.06506900 -0.71632600 H 3.96742400 2.74434900 0.60406700 C 1.56893400 -1.98107800 -0.29269300 C 1.30233100 -2.93000700 -1.30906200 C 1.09430700 -4.26593200 -0.98296100 C 1.17347000 -4.67251600 0.35559500 C 1.47016100 -3.75701900 1.37069600 C 1.68131300 -2.41801700 1.04990100 H 2.24754100 -0.83959600 -0.61778500 H 1.25117200 -2.60695300 -2.34513000 H 0.87491000 -4.99364300 -1.75911700 H 1.00878700 -5.71725300 0.60728700 H 1.53883100 -4.09186400 2.40183100 H 1.92730100 -1.69198800 1.81891600 C H O 0 6-31G* **** Pd 0 LanL2DZ **** Pd 0 LanL2DZ !所有空白行都是特意的,不要忽略 !注意:第3行的genecp表示这里使用赝势基组 |
图13. σ-复分解过渡态优化输入文件
图14为优化得到的过渡态结构:
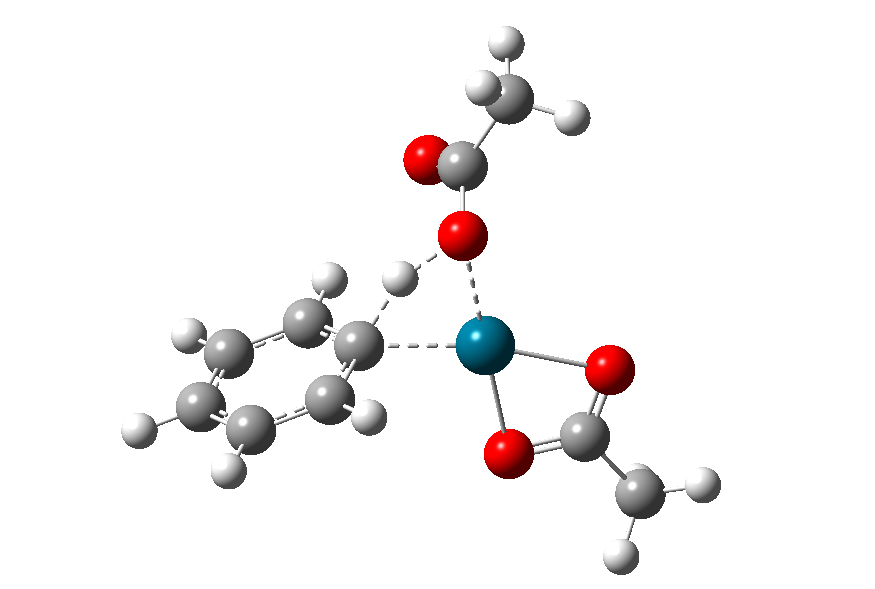
图14. σ-复分解过渡态优化得到的结构
三.总结
我们绘制了三种断键模式下的势能面(图15),可以看出,σ-复分解机理的路径(红色路径)有着远高于其他两种路径的能垒,在这个例子中,尽管我们看到外源碱辅助的去质子化路径有着最低的能垒(黑色路径),但在实际研究中,我们还需要考虑形成游离的布朗斯特碱所需要的能垒。为了方便说明,在这里,我们已经把配体类型尽可能做了简化,在研究中,通常还会加入其他的配体类型(比如N杂环配体),但基本的研究思路还是与我们的介绍一致。
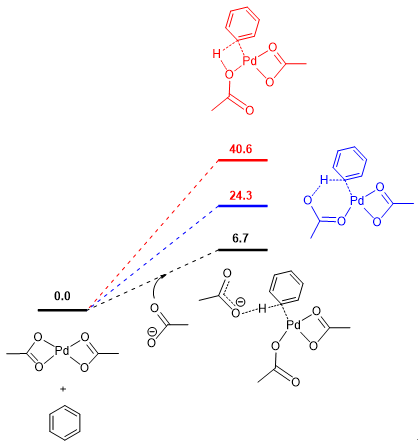
图15. 三种断键模式下的势能面
尽管我们的例子列举了3种C-H键断裂模式(对于其他的体系,还有不同类型的C-H键断裂模式),但我们在实际的研究中并不需要尝试所有的类型,因为前人的研究结果揭示了某些催化体系可能更偏爱特定类型的机理,但需要保持警惕是,这些经验并不是绝对的,还需要根据自己的研究体系找出最佳的反应机理,也就是说,当面对一个催化体系时,如果没有足够的证据证明它的催化模式,就需要多尝试几种断键模式。
需要注意的是,在这篇教程里,我们只计算了气相下分子的吉布斯自由能,在具体的研究中,反应通常是在溶剂中进行,这时为了得到更准确的反应能垒,需要计算分子的溶剂化自由能。


