
摘要:本文综述了分子指纹图谱的概念,描述了分子指纹图谱的不同方法,以及在虚拟筛选中的应用。
肖高铿/2019-01-29
[latexpage]
1.分子指纹(Molecular Fingerprint)
在比较两个化合物之间的相似性时遇到的最重要问题之一是任务的复杂性,这取决于分子表征的复杂性。 为了使分子的比较在计算上更容易,需要一定程度的简化或抽象。分子指纹就是一种分子的抽象表征,它将分子转化(编码)为一系列比特串(即比特向量,bit vector, 见Figure 1.),然后可以很容易地在分子之间进行比较。典型的流程是将提取分子的结构特征、然后哈希(Hashing)生成比特向量。

Figure 1. 比特向量
比较分子是很难的,比较比特串却很容易,分子之间的比较必须以可量化的方式进行。分子指纹上的每个比特位对应于一种分子片段(Figure 2),假设相似的分子之间必然有许多公共的片段,那么具有相似指纹的分子具有很大的概率在2D结构上也是相似的。
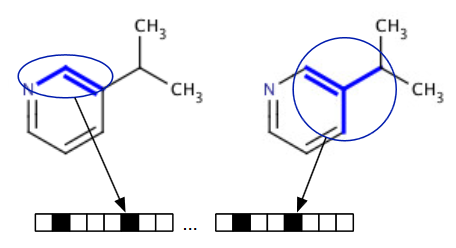
Figure 2. 比特向量上的每个位置对应一个分子片段或特征
有十多种方法可以评估两个向量之间的相似性(参见:Fingerprints-Screening and Similarity.),最常见的是欧几里德距离。但对于分子指纹,行业标准是Tanimoto系数,它由两个指纹中设置为1的公共位数除以两个指纹之间设置为1的总位数组成。这意味着Tanimoto系数总是具有介于1和0之间的值,而不管指纹的长度如何,这导致指纹随着指纹变长而变得松散。这种损失还意味着具有给定Tanimoto系数的两个指纹实际上将如何相似地将极大地取决于所使用的指纹的类型,这使得不可能选择用于确定两个指纹是相似还是不相似的通用截止标准。然而,通过数据融合策略将分子指纹与其他相似系数相结合,可以提高分子指纹的性能[1]。表1列出了几个与指纹一起使用的相似性和距离度量。
Table 1. 几种指纹的相似性和距离度量[10]
| 方法 | 表达式 | 取值范围 |
|---|---|---|
| Tanimoto/Jaccard coefficien | \[\frac{c}{a+b-c}\] | 0-1 |
| Euclidean distance | \[\sqrt{a+b-2c}\] | 0-N |
| City-block/Manhattan/Hamming distance | a+b-2c | 0-N |
| Dice coefficien | \[\frac{2c}{a+b}\] | 0-1 |
| Cosine similarity | \[\frac{c}{\sqrt{ab}}\] | 0-1 |
| Russell–RAO coefficien | \[\frac{c}{m}\] | 0-1 |
| Forbes coefficien | \[\frac{cm}{ab}\] | 0-1 |
| Soergel distance | \[\frac{a+b-2c}{a+b-c}\] | 0-1 |
其中,给定两个化合物A和B的指纹,m等于指纹中存在的总位数,a、b分别等于A、B中比特值为1的位数,c等于A和B中公共的比特值为1的位数。
1.1 分子指纹的类型
根据将分子表征转换成位串方法的不同,分子指纹可以分为几种类型。 大多数分子指纹的编码方法仅使用了2D的分子图形信息,因此称为2D指纹; 一些方法能够存储3D信息,比如是药效团指纹。常见的分子指纹方法包括基于子结构的指纹(substructure key-based fingerprint),基于拓扑或路径的指纹(topological or path-based fingerprint)和圆形指纹(circular fingerprint)。
1.1.1 基于子结构的指纹(substructure key-based fingerprint)
基于子结构的指纹根据给定结构列表中某些子结构或特征的存在与否来设置位串。 这意味着如果分子主要由指纹的子结构组成时最有用,而当分子包含指纹的子结构时则不那么多时则不会很有用,因为它们的特征将不被分子指纹所表示。 一种分子指纹的位数由子结构的数量决定,每个比特位与分子中单个给定特征的存在或不存在有关(Figure 3),这与其他(散列)类型的指纹不同。 最常用的基于子结构的分子指纹有:
- MACCS[2]
- PubChem指纹[3]
MACCS采用SMARTS编码的子结构,根据子结构种类数量不同有两个变种:一种是166,另一种是960。 较短的是最常用的,因为它的长度相对较小(仅166位),但涵盖了药物发现和虚拟筛选时的大多数感兴趣的化学特征。大部分软件中的MACCS指纹用的是短版本,而较长的960版本则很少见,大部分软件包不能计算。
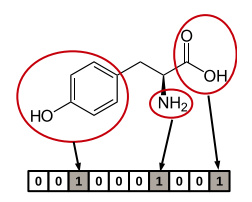
Figure 3. 一个10-BIT长度的指纹图谱示意图:每个位置编码一个子结构, 有3个位置所代表的子结构出现在上图的分子中(即被圆圈圈中的那些子结构),这些位置被标为1;其它的位置代表的子结构没有出现在分子中,则被标为0。
该指纹具有881个子结构,涵盖了广泛、多样的不同子结构和官能团。 PubChem将该指纹用于相似性搜索。 除了PubChem自己的代码之外,在PaDEL-Descriptor[4]和CDK[5,6]中都有实现。
1.1.2 基于拓扑或路径的指纹图谱(Topological or path-based fingerprint)
基于拓扑或路径的指纹通过分析从一个原子开始直至到达指定数量键的路径(通常为线性)上所有的分子片段,然后对每一个路径中的进行哈希(Hasing)产生指纹(Figure 4)。此类指纹适用于任意一个分子,并可以调整其长度,可以用于快速的子结构搜索与分子过滤。哈希的指纹意味从比特位出发无法追踪到结构特征。同样的比特位可以对映不同的结构特征,称为“比特位冲突”(bit collision)。Daylight指纹(Daylight fingerprint)[7]是此类型指纹中最突出的代表。它们由多达2048的比特位组成,编码了分子达到给定长度的所有可能的连接途径。大多数软件可实现此类指纹,有的软件可以达到更高的位数或使用非线性连接路径,例如OpenEye的Tree指纹(Tree fingerprint)[8]。
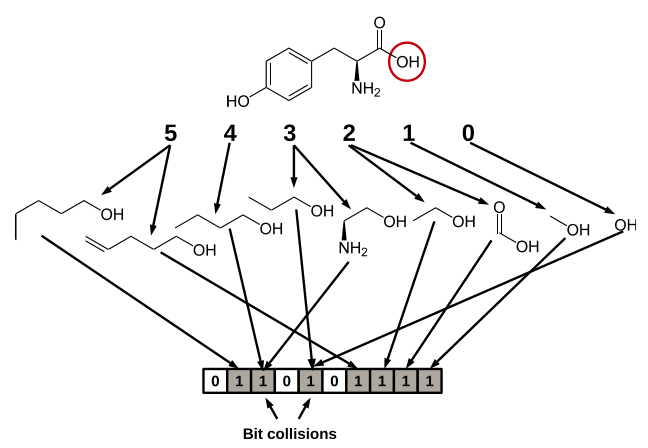
Figure 4. 采用线性路径分析长达5键路径上的所有分子片段生成的10比特位拓扑指纹。从起始原子(用圆圈标出)出发找到的所有片段,片段长度和相应的比特位用箭头指示。如图可见有两个比特位冲突,多个片段指向同一个比特位并引发比特位减少。上图仅展示了从一个单个起始原子出发的片段和比特位;对于完整的指纹,将对分子中的每个原子进行重复该过程。圆形指纹(Circular fingerprint)采用类似的方法,不同是的在起始原子特定半径内构建片段而不是线性路径上构建片段。
1.1.3 圆形指纹图谱(Circular fingerprints)
圆形指纹也是一种哈希的拓扑指纹,但它们与基于路径的指纹不同之处在于:不是在分子中寻找路径,而是记录每个从原子出发直到指定半径内的环境(见Figure 5)。 因此,此类指纹不适用于子结构查询(因为相同的片段可能具有不同的环境),但广泛用于完整结构的相似性搜索。
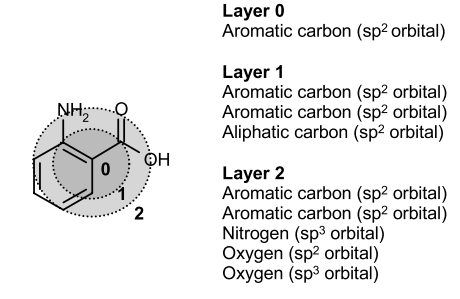
Figure 5. Circular Fingerprint示意图[9]:以一个重原子为中心,搜寻在特定半径范围(有的实现用直径)内的结构特征,比如力场的原子类型,官能团,片段等等各种信息。
- Molprint2D
- ECFP
- FCFP(Functional-Class Fingerprints)
MolPrint2D是由Bender等(2004)开发[11,12],常用于QSAR研究与比较分子的相似性。OpenBabel与jCompoundMapper提供了MolPrint2D指纹生成方法。
从Morgan算法[13]衍生出来的扩展连接指纹(Extended-Connectivity Fingerprints,ECFP)已经成为事实上圆形分子指纹的行业标准方法,专门设计用于构效关系研究[14]。 ECFP指纹在使用的时候,根据设定的直径不同会产生可变长度的指纹。最常用的是直径为4的ECFP4,还有直径为6的ECFP6,一些基准测试显示两者之间的性能差异很小[15]。 此外,还有一些变体比如ECFC还记录了ECFP特征的频率计数,而不仅只是是否出现。多种软件都提供了ECFP或Morgan算法,比如Pipe-line Pilot, Chemaxon的JChem, CDK和RDKit。注意,在RDkit中称为Morgan Fingerprint,路径长度用半径表示而不是直径,因此在RDkit的半径2相当与ECFP的直径4指纹。根据Gregory Landrum的测试,Morgan指纹与ECFP在相似性比较上没有显著差异[16]。
根据Rogers与Hahn的研究[14],一般来说,直径小的ECFP4足够适合于相似性搜索与分子聚类;而直径更大的ECFP得益于其包含更多的分子结构细节,因此适合于机器学习进行活性预测等,但限于计算量,通常用ECFP6与ECFP8。
FCFP是ECFP的一种变体,它进一步被抽象:它不是索引环境中的特定原子,而是索引该原子的作用。因此,具有相同或相似功能的不同原子或基团在该指纹里是没有区别的,这使它们可以作为一种药效团指纹。还有其它的FCFC变体,类似于ECFC对ECFP的变化。支持ECFP指纹的主要软件包基本也支持FCFP。
1.1.4 混合指纹(hybrid fingerprint)
有些分子指纹方法将不同指纹方法产生的比特串组合成为一个新的比特串,常用的包括:
- UNITY 2D
- MP-MFP
UNITY 2D是SYBYL软件包UNITY模块使用的一种指纹图谱,它组合了基于子结构与连接路径片段两种方法,长度为988比特位。
Ling Xue等人(Xue 2003)[17]设计开发的MP-MFP指纹包含了171比特位,其中110比特位编码化学子结构,61位编码了性质描述符(Figure 6)。

Figure 6.MP-MFP指纹示意图:浅灰色编码了性质信息(比如氢键受体、供体),深灰色编码了化学子结构信息。
1.1.5 药效团指纹图谱(Pharmacophore fingerprint)
药效团指纹也是常用的一种分子指纹。药效团代表了分子对给定靶标具有活性所需的相关特征和相互作用。 药效团指纹通常以类似于基于子结构的指纹方式编码分子的结构特征,但同时考虑了这些特征之间的距离,通常按距离范围对其进行分类生成比特位串。 这样,3D的药效团信息就可以编码到指纹中去。比如SYBYL软件包中的Tuplet就可以编码分子的3D药效团22(Figure 7)。在Tuplet中,分子的形状也用类似的方式编码,因此可以同时比较药效团与分子形状。
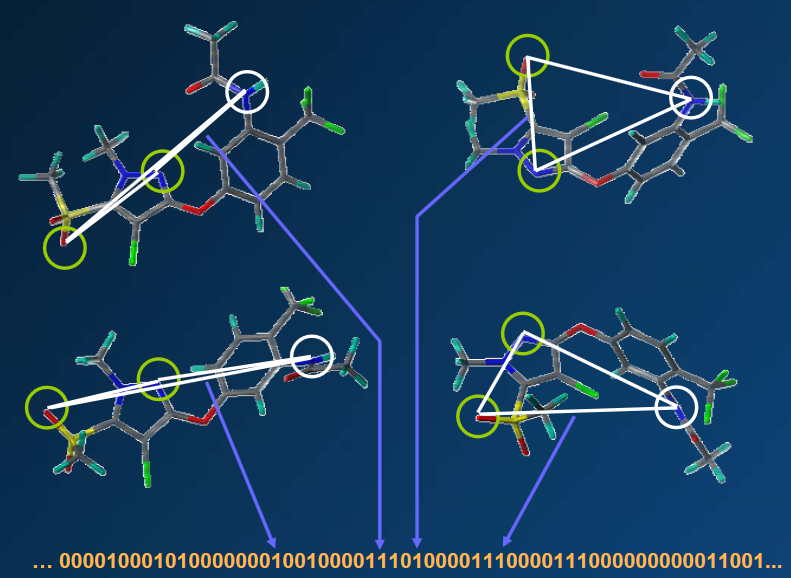
Figure 7. Tuplet药效团指纹图谱示意图:一个分子不同的构象,将每个构象的药效团特征按聚类分类、编码为比特串。
1.1.6 其它类型
LINGO [18]和SMIfp [19]是基于文本的分子指纹编码方法,这两种指纹从化合物的规范SMILES编码(canonical SMILES)出发生成化合物的指纹。分子指纹还可以基于结构的信息编码蛋白质-配体之间的相互作用,比如Da C等人(2014)[20]提出的蛋白-配体相互作用指纹(Structural Protein-Ligand Interaction Fingerprint,SPLIF)与Deng等人(2004)[21]提出结构相互作用指纹(Structural Protein–Ligand Interaction Fingerprints,SIFt)则编码了蛋白质-配体相互作用的信息,例如氢键、离子相互作用、与其残基的表面接触等等。
2. 基于指纹图谱虚拟筛选的软件
- OEChem TK/OpenEye
- JChem/ChemAxon
- Open Babel
- RDKit
- CDK
- Indigo
- Cinfony
- ChemFP
- Canvas/Schrodinger
- MOE
- jCompoundMapper
- Pipeline Pilot
- SYBYL-X
- FLAP
- MayaChemTools
OpenEye的OEChem TK可以生成166位的MACCS,LINGO,Circular,Path(与Daylight类似)和Tree(与Dayligth类似的非线性、“树”片段)指纹,提供了C ++,Java,Python和C#接口。
支持ECFP及其全部的变种ECFC, FCFP,FCFC以及药效团指纹图谱。
开源软件,支持MOLPRINT2D、166-bit MACCS、 Daylight样指纹FP2、FP3,提供了C++, Python、Perl、Ruby与Java等接口。
也是一款免费、开源的化学信息学工具包,支持多种指纹:MACCS(166-Bit)、与Daylight类似拓扑指纹、Atom Pairs( 分子中每个原子对基于原子环境和最短路径分离),Morgan指纹,Torsion指纹(基于拓扑两面角描述符)和Layered指纹。RDKit提供了C++,Python,Java以及C#等API。
支持ECFP, Estate, LINGO,MACCS, Daylight类似指纹图谱。
免费的开源化学信息学工具包,包含了几个哈希指纹及其组合,提供了C ++、Java、Python和C#等开发接口。
整合了Open Babel, RDKit, CDK, JChem与Indigo的全部功能。
用Open Babel, RDKit与OEChem做为后端进行计算。
MACCS,MOLPRINT2D, ECFP与线性路径指纹。
TGD, TGT,MACCS, 2D与3D的四点药效团指纹,EigenSpectrum形状指纹。
这是一款开源命令行软件,使用CDK支持多种指纹,包括MOLPRINT2D,原子对和药效团指纹等。 此外,它还提供了几种机器学习工具。
Pipeline Pilot本身是一款具有数据流工具,它可以计算各种指纹,包括MACCS、ECFP及其变体。
SYBYL-X采用其自己的UNITY 2D指纹进行数据库搜索。
FLAP是MolDiscovery开发是一种虚拟筛选软件,FLAP的指纹编码了分子的四点药效团特征信息,可用于配体-配体、配体-受体和受体-受体的比较。
MayaChemTools是用Perl写的程序,可以计算多种分子指纹,包括ECFP,MACCS,基于路径的指纹等等; 它也可以直接用于指纹的相似性搜索。
3.使用指纹进行虚拟筛选的场景
用指纹相似性进行虚拟筛选,需要具备以下条件:
- 至少一个已知的活性化合物做为参比分子(reference)
- 一个数据库,其中含有潜在的活性化合物
- 软件有能力生成并比较指纹
一旦确定了参比分子,下一步就是选择最合适的指纹。选择通常受限于软件的选项。最合适的选择还取决于参比分子,因为指纹应该能够正确地表征参比分子(哈希指纹不需要考虑这一点)。还应考虑数据库和可用指纹是否考虑立体化学、互变异构形式以及参比分子和数据库分子的构象。应优先使用立体化学敏感方法筛选立体化学敏感数据库。如果构象很重要的话,应该使用能够编码构象信息的指纹。还应考虑所研究分子的互变异构现象,因为同一分子的不同互变异构体可具有显着不同的指纹。
使用所选择的算法计算数据库中每个分子和参比分子的指纹,然后计算参比分子和数据库中每个分子之间的相似性系数。再根据相似系数对分子进行从大到小排序。最靠前的那些分子应该具有与参比分子相似的生物活性。
4. 与其它虚拟筛选方法的比较
4.1 3D方法优于指纹
Tresadern等人[23]在对CRF1受体进行虚拟筛选时比较了几种采用基于配体的方法:ECFP6指纹,特征树(Feature tree),Topomer,ROCS Shape Tanimoto,EON Electrostatic Tanimoto、OpenEye ComboScore(Shape Tanimoto和Color score的组合)和Cresset Fieldscreen。 结果表明:在4个计算中ECFP6指纹有3个表现最差,但ECFP6在其中一个计算中的性能优于其它方法。同时,3D方法可以也比2D指纹的方法发现结构更多样、更多骨架类型的化合物。
4.3 2D相似性方法优于3D相似性方法与分子对接
McGaughey等人[24]比较了几种拓扑、形状与分子对接等基于配体与基于结构的虚拟筛选方法。在她的研究中,2D相似性的比较用到了Daylight指纹与TOPOSIM,3D相似性测试了SQW与ROCS,分子对接测试了FLOG、FRED与Glide。在该研究中,作者考察了这些软件在11个靶标、2个化合物数据库(MDDR与Merck内部数据库)上的虚拟筛选性能。就多个靶标的平均富集因子而言,基于配体的方法要优于基于结构的方法(见Figure 8,9)。与其他多种虚拟筛选方法进行的比较,2D相似性方法优于大多数其他方法。作者得出结论:“就EF值而言,2D相似性方法(TOPOSIM,Daylight)在对具有结构多样性的数据库进行骨架跃迁时表现良好…”。
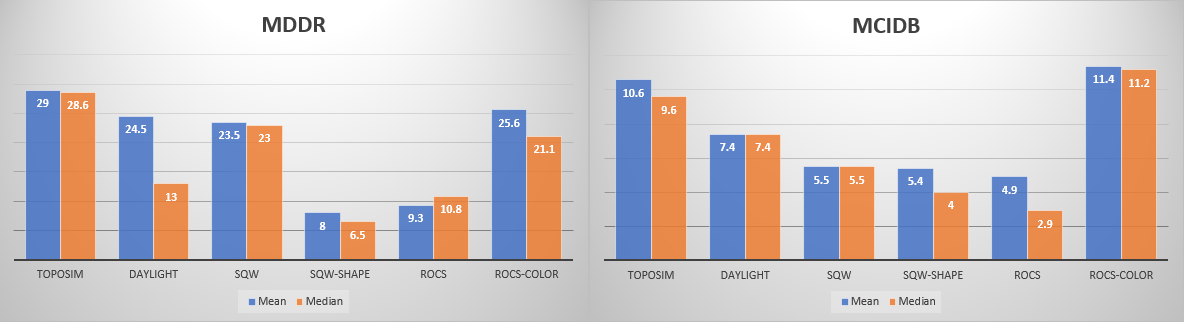
Figure 8. 基于配体的方法性能比较
从Figure 8还可以发现,用MDDR数据集评估的性能比用MCIDB数据集的要好。其中的一个原因可能是:MDDR文献数据居多,各个作者可能采用跟进策略因此公开的化合物具有更多的相似性,而使得虚拟筛选更容易;而MCIDB是Merck公司自己的数据,对化合物多样性要求更高,这使得虚拟筛选更难。
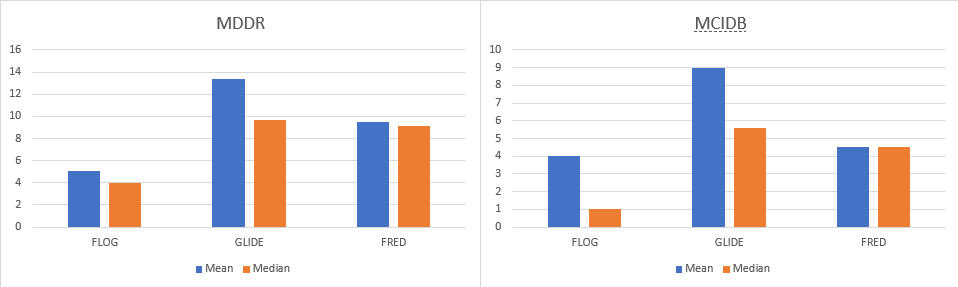
Figure 9. 基于结构的方法性能比较
同样的,对于基于结构的方法,从Figure 9也观察到MDDR数据集比MCIDB数据集更容易虚拟筛选。比较Figure 8与Figure 9,基于配体的方法富集因子远远地比基于结构的要高。
Venkatraman等人用DUD数据集比较了几种基于配体方法的虚拟筛选性能,作者将指纹(Open Babel FP2,BCI,MACCS,Daylight和MOLPRINT2D)与基于3D的分子形状的方法(ESHAPE3D,ROCS,PARAFIT,SHAEP和USR)进行了比较。作者总结到:“总的来说,我们发现基于2D指纹的方法比许多基于3D形状的方法在DUD数据集上表现出更好的虚拟筛选性能”。 这表明了3D方法并不总是优于简单的指纹相似性搜索。
文献
- Salim N, Holliday J, Willett P. Combination of Fingerprint-Based Similarity Coefficients Using Data Fusion. J Chem Inf Comput Sci. 2003;43(2):435-442. doi:10.1021/ci025596j.
- Durant, J. L.; Leland, B. A.; Henry, D. R.; Nourse, J. G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42 (6), 1273–1280. https://doi.org/10.1021/ci010132r.
- Cereto-Massagué A, Ojeda MJ, Valls C, Mulero M, Garcia-Vallvé S, Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods. 2015;71:58-63. doi:10.1016/j.ymeth.2014.08.005.
- Yap CW. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32(7):1466-1474. doi:10.1002/jcc.21707.
- CDK (Version 2.1), https://cdk.github.io
- Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J Chem Inf Comput Sci. 2003;43(2):493-500. doi:10.1021/ci025584y.
- Daylight chemical information systems, Daylight. http:// www.daylight.com. (accessed on 26/01/2019).
- OEChem,OpenEye scientific software. http:// www.eyesopen.com. (accessed on 26/01/2019).
- Glem, R. C.; Bender, A.; Arnby, C. H.; Carlsson, L.; Boyer, S.; Smith, J. Circular Fingerprints: Flexible Molecular Descriptors with Applications from Physical Chemistry to ADME. IDrugs 2006, 9 (3), 199–204. https://doi.org/16523386.
- Cereto-Massagué, A.; Ojeda, M. J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular Fingerprint Similarity Search in Virtual Screening. Methods 2015, 71, 58–63. https://doi.org/10.1016/j.ymeth.2014.08.005.
- Bender, A.; Mussa, H. Y.; Glen, R. C.; Reiling, S. Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naïve Bayesian Classifier. J. Chem. Inf. Comput. Sci. 2004, 44 (1), 170–178. https://doi.org/10.1021/ci034207y.
- Bender, A.; Mussa, H. Y.; Glen, R. C.; Reiling, S. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44 (5), 1708–1718. https://doi.org/10.1021/ci0498719.
- Morgan, H. L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5 (2), 107–113. https://doi.org/10.1021/c160017a018.
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742–754. https://doi.org/10.1021/ci100050t.
- Riniker, S.; Landrum, G. A. Open-Source Platform to Benchmark Fingerprints for Ligand-Based Virtual Screening. J. Cheminform. 2013, 5 (1), 26. https://doi.org/10.1186/1758-2946-5-26.
- Landrum, G. Fingerprints in the RDKit. RDKit UGM 2012 2012. http://rdkit.org/UGM/2012/Landrum_RDKit_UGM.Fingerprints.Final.pptx.pdf (accessed on 26/01/2019)
- Xue, L.; Godden, J. W.; Stahura, F. L.; Bajorath, J. Design and Evaluation of a Molecular Fingerprint Involving the Transformation of Property Descriptor Values into a Binary Classification Scheme. J. Chem. Inf. Comput. Sci. 2003. https://doi.org/10.1021/ci030285+.
- Vidal, D.; Thormann, M.; Pons, M. LINGO, an Efficient Holographic Text Based Method to Calculate Biophysical Properties and Intermolecular Similarities. J. Chem. Inf. Model. 2005. https://doi.org/10.1021/ci0496797.
- Schwartz, J.; Awale, M.; Reymond, J.-L. SMIfp (SMILES Fingerprint) Chemical Space for Virtual Screening and Visualization of Large Databases of Organic Molecules. J. Chem. Inf. Model. 2013, 53 (8), 1979–1989. https://doi.org/10.1021/ci400206h.
- Da, C.; Kireev, D. Structural Protein–Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. J. Chem. Inf. Model. 2014, 54 (9), 2555–2561. https://doi.org/10.1021/ci500319f.
- Deng, Z.; Chuaqui, C.; Singh, J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein−Ligand Binding Interactions. J. Med. Chem. 2004, 47 (2), 337–344. https://doi.org/10.1021/jm030331x.
- Fox, P. C.; Wolohan, P. R. N.; Abrahamian, E.; Clark, R. D. Parameterization and Conformational Sampling Effects in Pharmacophore Multiplet Searching. J. Chem. Inf. Model. 2008, 48 (12), 2326–2334. https://doi.org/10.1021/ci800234q.
- Tresadern, G.; Bemporad, D.; Howe, T. A Comparison of Ligand Based Virtual Screening Methods and Application to Corticotropin Releasing Factor 1 Receptor. J. Mol. Graph. Model. 2009, 27 (8), 860–870. https://doi.org/10.1016/j.jmgm.2009.01.003.
- McGaughey, G. B.; Sheridan, R. P.; Bayly, C. I.; Culberson, J. C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.-F. F.; Cornell, W. D. Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47 (4), 1504–1519. https://doi.org/10.1021/ci700052x.
- Venkatraman, V.; Pérez-Nueno, V. I.; Mavridis, L.; Ritchie, D. W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools Against the DUD Data Set Reveals Limitations of Current 3D Methods. J. Chem. Inf. Model. 2010, 50 (12), 2079–2093. https://doi.org/10.1021/ci100263p.


