
摘要:本文叙述了拜尔公司科研人员用SPARK发现高效、选择性的SOS1抑制剂的研究过程:首先用片段筛选与高通量筛选发现了与SOS1结合的片段F1与化合物17;它们分别结合于SOS1的子结合口袋与主结合口袋;F1与17有部分重合,删除该重合后用Spark合理地设计了连接F1与17的连接臂;最后发现了化合物BAY-293,是一种KRAS-SOS1相互作用强效、选择性的抑制剂;并与KRAS的共价抑制剂具有协同作用。
编译:肖高铿
时间:2019-06-25
前言
GTPase的RAS家族由KRAS、NRAS与HRAS等组成,它们是主要的致癌基因,在人类癌症中的发生率高达20-30%1-3。RAS蛋白是个分子开关,在GTP结合的活性态与GDP结合的非活性态之间互相切换。在鸟嘌呤核苷酸交换因子(Guanine nucleotide exchangge factor,GEF)的激活下,处于GTP结合状态的RAS与许多效应物(effector)相互作用。 GTPase激活蛋白(GAPase-activating proteins, GAPs)通过加速内源性弱GTPase活性来下调活性RAS(高达5个数量级),从而驱动RAS恢复到失活状态。然而,致癌RAS突变体的GAP活性受损或大大降低,导致其被永久活化,这是致癌RAS信号传导的基础4;例如,通过RAS-RAF-MEK-ERK和RAS-pi3k-PDK1-AKT通路,两者都是细胞存活和增殖所必需的5。
由于GTP对RAS结合位点具有极高的结合亲和力(皮摩尔水平)、并且结合位点缺乏其明确界定的口袋、以及RAS与GEF、GAP和效应物之间的蛋白-蛋白相互界面宽阔且平坦,这些都导致开发RAS的直接抑制极具挑战性。此外,试图开发非直接抑制剂也困难重重,比如靶向法尼基转移酶(Farnesyl transferase)间接抑制RAS的尝试尚未得到批准的药物6。鉴于这些直接或间接的方法目前都没获得成功的药物,RAS一度被认为是不可成药靶标。目前直接抑制RAS的策略主要聚焦于:
- 用共价抑制剂靶向致癌突变体KRASG12C的Cys12
- 靶向RAS-效应子相互作用(RAS-effector interaction)以干扰下调信号
- 抑制RAS-GEF相互作用以阻止重新载入GTP7
前两个策略已经取得令人鼓舞的成功8-11,而靶向RAS-GEF相互作用目前尚未发现强效的抑制剂。此外,突变的RAS蛋白是否需要GEF活性来完全激活仍有待充分研究,并且可能因具体突变而异。研究最多的GEF是蛋白质SOS(Son of Sevenless),已知其有两种亚型SOS1和SOS2。人们已经对正构SOS螺旋设计模拟肽(peptide mimicking)来抑制RAS-SOS相互作用,发现了具有纳摩尔级别结合亲和力的碳氢订书肽(hydrocarbon-stapled peptide),遗憾的是,它的细胞活性很低12-13。采用基于片段的筛选、理性设计与高通量筛选也发现了靶向KRAS-SOS1相互作用、具有中等微摩尔亲和力的小分子14-17。令人吃惊的是,一些配体激活了SOS1介导的核苷酸交换而不是抑制,从而通过对SOS1的负反馈导致RAS信号的双相调节18。
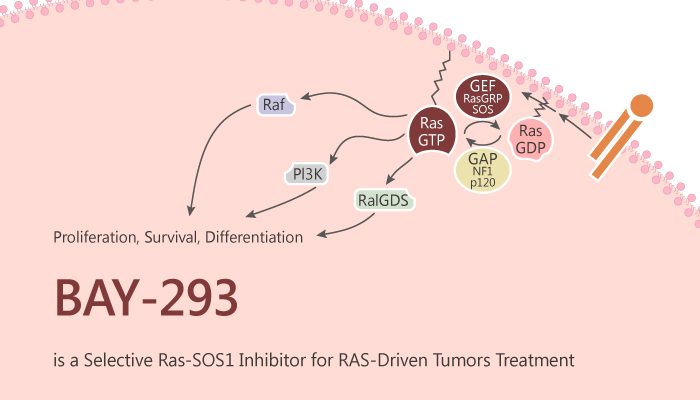
拜尔公司的Roman C. Hillig等人19最近报道了高效、选择性抑制SOS1激活KRAS的小分子抑制剂BAY-293,该抑制剂能够通过干扰RAS-SOS1相互作用而阻断RAS活化。因致癌基因KRASG12C突变体在肺癌中的重要性而受到研究者的关注20,在该文中,研究人员首先通过片段筛选与高通量筛选分别发现了片段分子F1与SOS1抑制剂化合物17。高通量筛选命中的化合物17结合于SOS1的主结合口袋(PDB:5OVF),而分子片段F1结合于主结合口袋旁边的子结合口袋(PDB:6EPM)。F1与化合物17有部分叠合,然后作者将两者的叠合部分删除,用Spark的分子连接策略进行了虚拟筛选,合理地设计了可以连接F1与化合物17的连接臂,最后发现了化合物BAY-293,是一种KRAS-SOS1相互作用的强效、选择性的抑制剂。我们对该文的分子设计部分进行了综述,与大家分享该文关键的设计方法与结论。
基于片段的筛选
拜耳的科研人员决定采取双管齐下的方法:同时开展高通量筛选与和片段筛选。对一个含有3,000个片段的化合物库筛选出了与KRAS-SOS1蛋白-蛋白相互作用位点结合并可在该位点诱导构象变化的片段。其通过触发Phe890侧链的旋转,然后打开了与主结合口袋相邻的新子结合口袋。
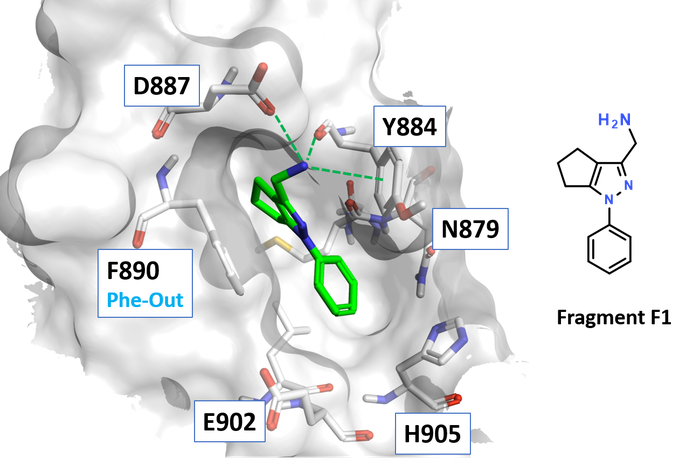
Figure 1. F1-SOS1复合物晶体结构(PDB: 6EPM)
片段F1被选作为进一步优化的起点。 F1与SOS1复合物晶体结构(pdb:6epm,图1)表明苯基部分与Phe890(Phe-out)发生 π-π 相互作用。氨甲基部分与Asp887和Tyr884的主链羰基形成氢键,并与Tyr884侧链形成额外的阳离子-π 相互作用。
高通量筛选与初步的优化
拜尔的科研人员还对一个含300万化合物的数据库进行了高通量筛选,结果发现了化合物1(IC50=320nM,Figure 2.)
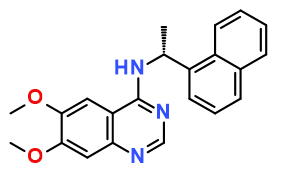
Figure 2. 化合物1(IC50=320nM)
用吡唑基苯基取代化合物1中的萘基部分得到化合物17(图3),显示出对SOS1的良好活性(IC50 = 140nM)和改善的水溶性。
化合物17与SOS1复合物晶体结构(pdb:5ovf,图3)解释了作用模式:化合物17的喹唑啉骨架夹在His905和Tyr884之间(π-π 堆叠)。吡唑基苯基部分占据了由Leu901和Phe890(Phe-in)组成的疏水口袋中,并与Tyr884的侧链有T-堆积相互作用。吡唑部分与Glu902形成水桥连的H-键。中心苯胺NH与Asn879的侧链形成H-键。
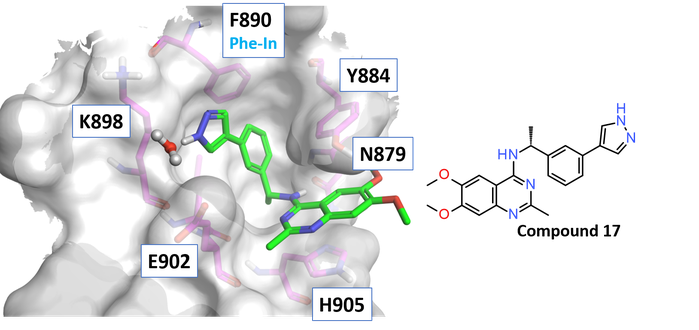
Figure 3: 化合物17与SOS1SB的复合物晶体结构(PDB:5OVF)
用SPARK的分子连接策略将片段F1与化合物17的连接起来
由于片段筛选识别了尚未被高通量筛选命中化合物结合的新的子口袋,拜尔的科研人员尝试了使用Spark的配体连接方法:通过组合两种配体来进一步提高活性。如图4-A分析了化合物17(pdb:5ovf)和F1(pdb:6epm)晶体结构的叠加效果,表明这个策略是可行的。
将化合物17和F1叠合的芳基切除,作者用Spark进行了配体连接实验(图4-b,c)以确定合适的连接臂:使F1的四氢环戊[c]吡唑部分和17的氨基喹唑啉基团在SOS1的活性部位正确定位。在Spark给出的得分最高的连接臂中,噻吩连接臂被选中、合成、最后生物学测试发现是活性的。
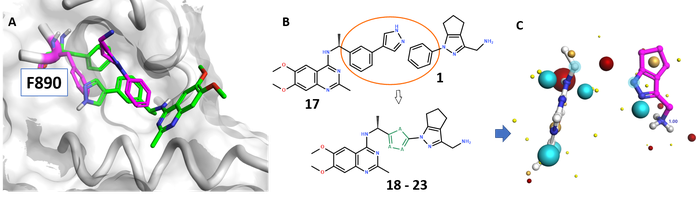
Figure 4. 片段F1与化合物17的配体连接: Figure 4A为F1-SOS1复合物结构(SOS1灰色, F1与Phe890洋红色)以及化合物17与SOS1复合物结构 (17与Phe890绿色); Figure 4B为片段连接策略示意图; Figure 4C为F1与17的场点图。
视频演示:起始结构的准备
本视频包含了:SOS1与F1、化合物17复合物结构的下载;蛋白结构准备;序列比对、蛋白叠合、化合物导出保存为起始结构starter.sdf。
视频演示:分子连接、发现先导化合物
在本视频中,包含了:(1)读入上一步准备好的起始结构;(2)准备连接点;(3)虚拟筛选。其中第一轮仅对对非常常见的片段库进行筛选,仅命中两个化合物。因此又进行了第二轮筛选,发现了文献报道的噻吩骨架:不同位置的取代基均被命中, 见Figure 5。
SPARK虚拟筛选命中的部分化合物如图5所示。其中左上角化合物为两个起始的分子;另外三个化合物为命中的化合物。
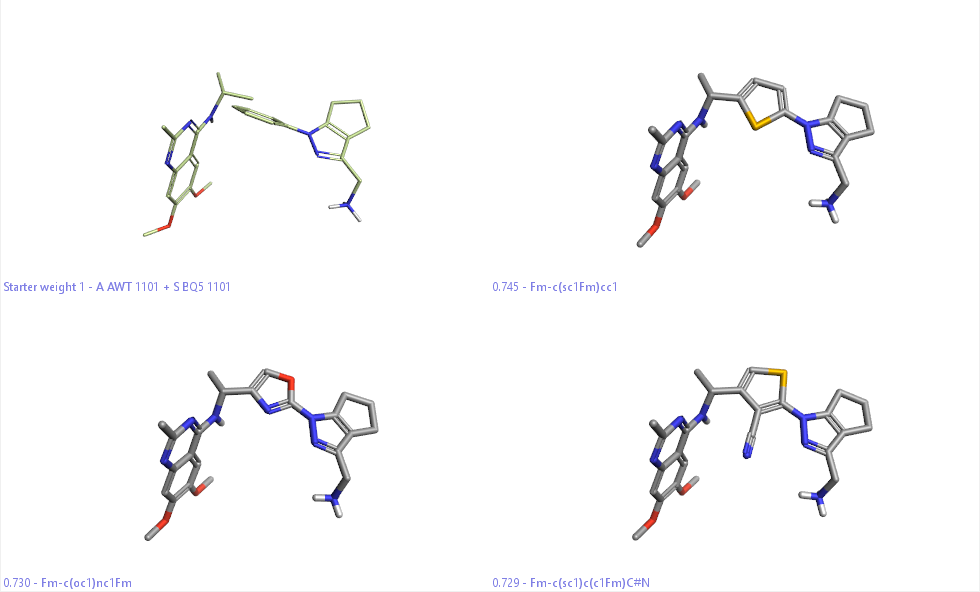
Figure 5.SPARK虚拟筛选结果示例。
进一步优化发现BAY-293
对噻吩连接臂杂交出的苗头化合物引入模拟F1片段与氨基侧链氢键相互作用的极性部分(如OH,NH2)可以触发Phe-out构象,导致发现了活性优化的化合物23(BAY-293,IC50=21nM)。 如图6所示,化合物23的侧链氨基通过与Asp887和Tyr884主链羰基形成H-键相互作用以及与Tyr884侧链的阳离子-π相互作用与SOS1相互作用。
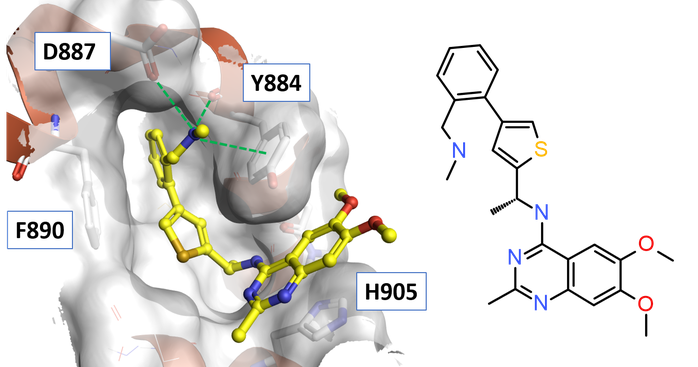
Figure 6.化合物23与SOS1结合位点的相互作用模式(PDB: 5OVI)
进一步的筛选和抗增殖数据证明化合物23(BAY-293)是靶向KRAS-SOS1相互作用有效、选择性的抑制剂,并且表明抑制GEF可能代表着靶向RAS驱动的肿瘤治疗可行方法。
结论
拜耳的这篇论文展示了Spark方法的实用性。 除了作为一个极好的生物电子等排体基团替换之外,Spark的先进功能还包括易于使用的水分子替换方法、分子大环化、分子生长和分子连接等等。在拜尔的这个研究中,Spark建议了如何将占据蛋白结合位点不同部分的两个不同分子连接起来,并将SAR从一个系列转移到另一个系列,SPARK在其中起了至关重要的作用。
文献
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014) Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13:828–851.
- Hobbs GA, Der CJ, Rossman KL (2016) RAS isoforms and mutations in cancer at a glance. J Cell Sci 129:1287–1292.
- Simanshu DK, Nissley DV, McCormick F (2017) RAS proteins and their regulators in human disease. Cell 170:17–33.
- Haigis KM (2017) KRAS alleles: The devil is in the detail. Trends Cancer 3:686–697
- Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22.
- Berndt N, Hamilton AD, Sebti SM (2011) Targeting protein prenylation for cancer therapy. Nat Rev Cancer 11:775–791.
- Ostrem JM, Shokat KM (2016) Direct small-molecule inhibitors of KRAS: From struc- tural insights to mechanism-based design. Nat Rev Drug Discov 15:771–785.
- Janes MR, et al. (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172:578–589.e17.
- Lito P, Solomon M, Li LS, Hansen R, Rosen N (2016) Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351:604–608.
- Patricelli MP, et al. (2016) Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 6:316–329.
- Sautier B, Nising CF, Wortmann L (2016) Latest advances towards ras inhibition: A medicinal chemistry perspective. Angew Chem Int Ed Engl 55:15982–15988.
- Patgiri A, Yadav KK, Arora PS, Bar-Sagi D (2011) An orthosteric inhibitor of the Ras- Sos interaction. Nat Chem Biol 7:585–587.
- Leshchiner ES, et al. (2015) Direct inhibition of oncogenic KRAS by hydrocarbon- stapled SOS1 helices. Proc Natl Acad Sci USA 112:1761–1766.
- Winter JJ, et al. (2015) Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. J Med Chem 58:2265–2274.
- Evelyn CR, et al. (2014) Rational design of small molecule inhibitors targeting the Ras GEF, SOS1. Chem Biol 21:1618–1628
- Burns MC, et al. (2018) High-throughput screening identifies small molecules that bind to the RAS:SOS:RAS complex and perturb RAS signaling. Anal Biochem 548: 44–52.
- Burns MC, et al. (2014) Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc Natl Acad Sci USA 111:3401–3406.
- Howes JE, et al. (2018) Small molecule-mediated activation of RAS elicits biphasic modulation of phospho-ERK levels that are regulated through negative feedback on SOS1. Mol Cancer Ther 17:1051–1060.
- Hillig RC, Sautier B, Schroeder J, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc Natl Acad Sci. 2019;116(7):2551-2560. doi:10.1073/pnas.1812963116
- Stephen AG, Esposito D, Bagni RK, McCormick F (2014) Dragging ras back in the ring. Cancer Cell 25:272–281.


