
使用增强采样沿着正则模坐标来探索隐秘口袋——KRASG12D案例研究
摘要:隐秘口袋的识别为开辟新的治疗机遇提供了可能——这类结合位点在靶蛋白的静态未结合配体蛋白构象(apo structure)中通常处于隐匿状态。此外,当天然配体结合位点在蛋白质变体间高度保守时,变构隐秘口袋可成为设计靶点选择性配体的关键突破口。以KRAS为例:在其变构隐秘口袋被发现前,该靶点因蛋白表面平滑、核苷酸结合位点(GDP/GTP)在野生型与致癌亚型中高度保守,长期被视为“不可成药”。近期在 KRASG12C突变体 中鉴定出的 Switch-II隐秘口袋(cryptic pocket),以及FDA批准靶向该位点的抗癌药物(如Sotorasib),凸显了隐秘口袋在破解“不可成药”靶点难题中的关键价值。在此,我们提出一种基于 加权系综分子动力学模拟(weighted ensemble molecular dynamics simulations,WE MD)的新方法,通过将 固有正则模式(inherent normal modes)作为进度坐标(progress coordinates),系统探索野生型KRAS与G12D突变体的隐秘口袋动态。本研究通过全原子分子动力学模拟,总时长\(>400 μs\),系统探索了KRAS蛋白在添加共溶剂(氙气/乙醇/苯)与无溶剂条件下的构象动力学。通过三种独立的轨迹分析方法(暴露子分析、动态探针结合分析、探针图分析),成功预测了KRAS中已知的隐秘口袋(cryptic pockets),验证了方法的普适性。在此基础上,进一步模拟了抑制剂MRTX1133与KRASG12D突变体的结合过程,揭示了隐秘口袋的动态形成机制,并明确了构象选择(conformational selection)与诱导契合(induced-fit)在口袋开放中的协同作用。
原文:Vithani, N. et al. (2024) “Exploration of Cryptic Pockets Using Enhanced Sampling Along Normal Modes: A Case Study of KRAS G12D,” Journal of Chemical Information and Modeling, 64(21), pp. 8258–8273. Available at: https://doi.org/10.1021/acs.jcim.4c01435.
大纲
引言
潜在配体结合位点的识别是基于结构药物发现的先决条件。蛋白质的构象采样可能揭示短暂开放但仍具成药性的结合位点,这些位点在未结合配体的蛋白(apo structure)静态实验结构中通常处于隐藏状态。此类结合位点被恰当地称为”隐秘口袋”,它们不仅为长期被视为“不可成药”的蛋白质开辟了药物发现新途径,还能在具有已知正构配体的靶标中揭示新的”变构位点”。因此,隐秘口袋的识别能够拓展可成药位点的范围,并扩大可用于治疗开发的”可靶向蛋白质”谱系。此外,正构位点可能难以靶向,原因在于其对天然底物具有高亲和力,且由于口袋序列在功能相似蛋白质中的保守性,靶向该位点可能增加”脱靶效应”风险。”变构隐秘口袋”的识别已被证明在设计针对具有挑战性的癌症靶标(包括KRAS的多种致癌突变体)的选择性抑制剂方面至关重要1−4。
KRAS属于一个相关的GTP酶家族,参与RAS-MAPK信号通路,该通路对细胞增殖、分化和存活至关重要。由于其在细胞周期中的深度嵌入,KRAS中的功能获得性突变约占所有人类癌症的14%。在这些突变中,80%是编码野生型KRAS基因中甘氨酸的第12密码子的单碱基替换6,7。KRAS对其典型配体GTP/GDP具有皮摩尔亲和力,因此开发靶向正构位点的竞争性抑制剂一直是药物发现的一个不可行策略5,8。KRAS的小尺寸和平坦表面为变构抑制剂设计带来了额外的障碍。因此,自20世纪60年代末发现以来,KRAS一直是抗癌治疗里一个难以攻克的靶标9。
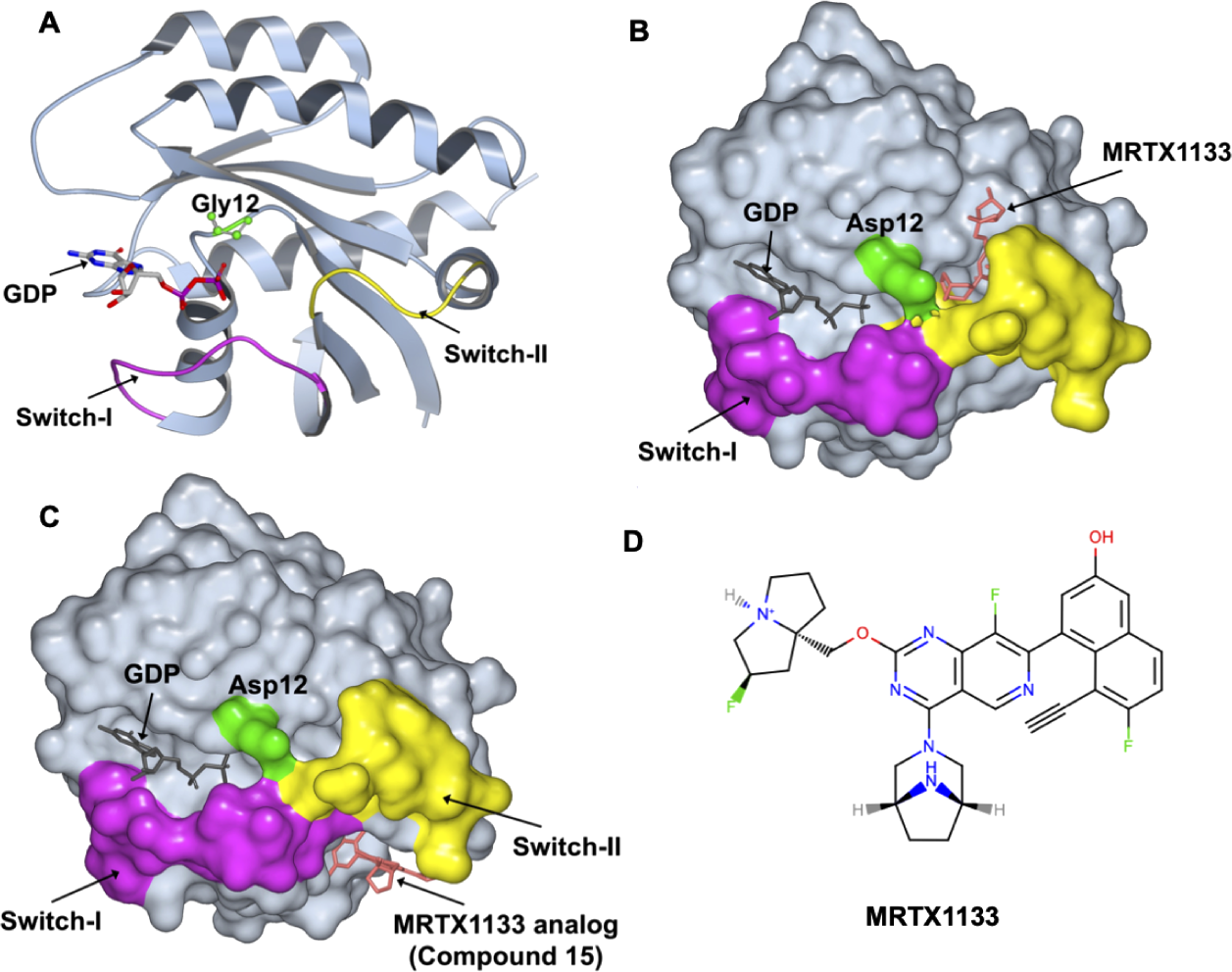
图1. KRAS:GDP结构,展示隐蔽口袋形成的关键区域。 (A) KRASWT:GDP复合物的晶体结构(PDB ID: 4OBE),其中开关I区(Switch-I)和开关II区(Switch-II)分别以品红色和黄色高亮显示。甘氨酸12残基(Gly12,与超过10%的人类癌症致癌突变相关)以绿色球棍模型呈现。(B) KRASG12D:GDP复合物的晶体结构以表面模型展示,MRTX1133结合于开关II口袋(Switch-II pocket)(PDB ID: 7RPZ),该口袋也是两种FDA批准药物(Sotorasib和Adagrasib)的结合位点。(C) 与MRTX1133类似物(化合物15)结合于开关I/II口袋(Switch-I/II pocket)的KRASG12D:GDP复合物晶体结构,以表面模型展示(PDB ID: 7RT1)。(D) 展示高效KRASG12D抑制剂MRTX1133的二维化学结构,该化合物结合于开关II口袋,并在本研究中使用。
2013年Shokat实验室进行的共价片段筛选研究取得突破,该研究表明KRASG12C的Switch-II区域附近存在一个变构隐秘口袋10。基于Shokat的原始发现,已经开发出靶向致癌突变体(KRASG12C和KRASG12D)中Switch-II隐秘口袋的选择性抗癌疗法(图1B)。比如Amgen(Sotorasib)11和Mirati Therapeutics(Adagrasib)12的FDA批准的用于KRASG12C的共价药物,以及Mirati Therapeutics开发的用于KRASG12D的I期临床试验(MRTX1133)中的非共价抑制剂13,以及Boehringer Ingelheim与Lito实验室合作开发的高亲和力泛KRAS非共价抑制剂(BI-2865)14(已知抑制剂的详细列表见表S1)。另外两个独立的片段筛选研究还发现了一个在Switch-I和Switch-II区域之间形成的额外隐秘口袋(图1C)15,16。已经设计出靶向这个Switch-I/II口袋的抑制剂来抑制SOS介导的KRAS和HRAS激活(详细列表见表S2)15−18。最近还在Switch-I/II区域附近报道了另一个口袋A59,该抑制剂结合复合物的晶体结构尚不可获得19。上述针对KRAS的抗癌药物发现的加速证明了变构(隐秘)口袋检测在基于结构的药物设计中的重要性。
KRAS抑制剂设计的进展说明了隐秘口袋的识别如何成为药物发现过程中的关键步骤。然而,寻找隐秘口袋在很大程度上仍然是偶然的,因为这样的位点只能通过在结合配体或片段存在的情况下解析蛋白质结构来实验检测。没有关于潜在配体集进行筛选的先验知识,隐秘口袋检测需要探索大量的蛋白质构象空间以揭示潜在的结合位点。分子动力学(MD)模拟已被证明是进行这种构象探索的强大计算工具。然而,隐秘口袋的形成可能发生在微秒或更长的时间尺度上,这在很大程度上是无偏全原子MD模拟无法达到的。虽然长时间尺度的MD模拟已用于隐秘口袋检测21,22,但增强采样方法,如副本交换23和无偏目标导向自适应模拟24,可以更有效地加速构象搜索。加权系综25,26(Weighted Ensemble,WE)MD模拟是另一种方法,可用于有效地增强沿(进度)坐标的构象采样,该坐标抽象地定义了隐秘口袋开放的机制。事实上,人们已经将WE MD算法(SubPEx)用于生成已知隐秘位点的子口袋的独特、类似平衡的构象27。在SubPEx中,WE进度坐标是使用感兴趣口袋位置和组成的现有知识来定义的。这些信息对于先前未探索蛋白质中隐秘口袋的前瞻性研究是不可用的。
从静态结构(如X-衍射、冷冻电镜或AlphaFold预测结构)通常难以揭示与隐秘口袋打开相关的构象动态及结构动力学信息,因而难以确立一个通用的进程坐标来有效增强此类运动的采样。然而,所有蛋白质均具备明确的集体运动(collective motions),即其固有动力学(intrinsic dynamics),这些运动通常表现为结构域的相对移动或构象的变构转变。正则模分析(Normal Mode Analysis, NMA)可有效估算这类全局动力学特征,因此为解析与口袋打开相关的大尺度构象变化提供了理论基础28,29。尽管已有研究通过将蛋白质结构沿选定的正则模式投影生成构象系综,并用于隐秘口袋的搜索30,但此类方法在采样微秒甚至更长时间尺度上发生的罕见事件(如隐秘口袋的形成)时可能受限。为此,诸如MDeNM等先进方法应运而生,它们通过向正则模式坐标注入动能,显著加速了这些稀有事件的动力学采样31。然而,另一种更具吸引力的策略是利用正则模式作为进展坐标以加速构象空间探索,同时避免对体系热力学或动力学本质的扰动。本研究提出,通过采样全局固有动力学而非局限于特定隐秘位点的局部运动,可实现无偏的构象采样。我们将正则模式系统性地整合至加权系综(Weighted Ensemble, WE)模拟框架中,作为通用的进展坐标,从而高效驱动大规模构象转变,有望揭示隐秘口袋及其他潜在功能相关的构象状态。
混合溶剂MD模拟也可以促进潜在配体结合位点的识别32。虽然纯溶剂模拟的结构波动可用来揭示可能打开的(子)口袋,但混合溶剂模拟提供了两个额外的见解:(1)共溶剂的占据和停留时间可以解释为给定位点潜在可成药性的度量,以及(2)共溶剂在子口袋的结合可能诱导蛋白质构象变化,通过诱导契合机制加速隐秘口袋的形成。常用的共溶剂包括苯、乙醇、丙酮、乙腈和其他小有机分子33−37。这些探针分子的类药特性使它们成为合适的共溶剂,然而,它们偏好的结合模式和结合位点特异性由其大小和化学特征(即形状、静电)决定,这可能会使隐秘口袋识别的结果产生偏差。为了规避这个问题,我们决定使用氙作为共溶剂,因为它的小尺寸、在水中的快速扩散以及以非特异性方式占据疏水位点的倾向38−48。虽然这对于隐秘口袋检测来说似乎是一个非常规的选择,但氙已被用于结构研究以定位被包埋的疏水腔49−51。尽管氙是一种化学惰性气体,但有证据表明其与细胞质和膜蛋白的结合是其作为全身麻醉剂38−43,47和神经保护剂45,48的药理作用的原因。氙还抑制NMDA和AMPA受体,它似乎作用于几种离子通道,如神经元烟碱乙酰胆碱(nACh)39,47和5-羟色胺3型(5-HT3)受体43,并且还显示出激活钾通道,包括双孔域钾通道(TREK-1)44和质膜ATP敏感通道(KATP)45。事实上,在一项晶体学研究中,氙被证明占据多种配体结合位点,包括溶剂可及的底物结合位点49。虽然对于所有的这些受体,确切机制细节有些神秘,但众所周知,氙通过直接结合蛋白质并改变其构象分布作用于一系列药理学相关靶标38。考虑到临床意义、在溶液中的高扩散率以及改变大蛋白质构象景观的能力,我们相信氙可能成为任意蛋白质复合物(包括KRAS)中隐秘位点的合理通用探针。
除了构象采样的挑战外,从生成的海量模拟数据中检测隐秘口袋还需要另一种复杂的计算方法。Bowman实验室开发的暴露子(exposon)分析方法通过找到经历溶剂可及性集体变化导致隐秘口袋关闭和打开的残基组来完成这项任务52。另一种流行的方法使用混合溶剂MD模拟进行隐秘口袋的检测,称为探针图分析(probe map analysis)33−37,53,涉及评估蛋白质表面上的共溶剂占据和结合自由能。在这项工作中,我们结合了暴露子和探针图分析方法,并提出了一种受暴露子方法学启发的新算法,称为动态探针结合分析。这种新的分析方法通过观察共溶剂/探针分子的集体结合行为来预测隐秘口袋。
在此,我们结合使用暴露子(exposon)、动态探针结合(dynamic probe binding)及探针图谱分析(probe map analysis)方法,系统分析了在纯水、两种共溶剂以及高活性类药分子存在条件下生成的超过400 μs的加权系综(Weighted Ensemble, WE)轨迹数据,旨在识别KRASG12D中的隐秘口袋。所提出的模拟与分析流程成功揭示了两个已知的配体结合位点,其中包括仅被两种突变选择性FDA批准药物靶向的关键口袋。为进一步深入理解KRASG12D中隐秘口袋形成的分子机制,我们还开展了MRTX1133结合过程的直接WE MD,以解析Switch-II口袋的动态形成路径。基于活性位点残基在形状与静电特征上的比较分析,这些模拟使我们能够明确区分该口袋形成过程中“构象采样”与“诱导契合”两种机制的相对贡献。尽管模拟中观测到的口袋构象与已报道的KRASG12D-抑制剂复合物晶体结构之间存在一定程度的差异,但本研究结果强烈提示:在Switch-II口袋的形成过程中,诱导契合机制可能起主导作用。
本文所提出的隐秘口袋检测流程构建了一个整合加权分子动力学(WE MD)与基于物理的分析方法的自动化框架,用于识别潜在的配体结合位点。此外,针对KRASG12D中MRTX1133结合过程的模拟揭示了预测隐秘口袋形成机制所面临的挑战:该过程可能依赖于特定化合物诱导的“诱导契合”机制,以实现配体与口袋之间精确的构象匹配。
方法
体系准备和MD模拟
所有蛋白质体系均首先通过OpenEye Spruce Toolkit54进行预处理:该工具以静态X-衍射晶体结构为输入,将生物大分子复合物从不对称单元扩展至其生物学组装体,补全缺失的残基和侧链,并添加氢原子;同时基于全局氢键网络的优化,生成一组能量有利的质子化态与互变异构体状态。随后,利用Orion云计算平台55中的MD Orion floe(工作流)对Spruce处理后的蛋白质结构进行自动化下游模拟准备。首先,将蛋白体系置于包含TIP3P水分子56的立方盒中,溶剂边界在各方向上均距蛋白表面至少10 Å。接着,添加适量Na⁺或Cl⁻离子以中和系统电荷,并额外加入150 mM NaCl以模拟生理盐浓度。在MD Orion floe内部,采用PACKMOL57生成多组分体系的初始构型。对于混合溶剂模拟(氙、乙醇、苯),在溶液相中随机引入150 mM的共溶剂/探针分子,并采用标准平衡流程进行初步弛豫。
体系参数化,蛋白质部分采用AMBER14SB力场58,小分子配体则使用OpenFF 2.0.0力场59。氙原子的参数仅包含Lennard-Jones势项(σ = 0.4063 nm,ε = 2.35 kJ/mol),其值来源于已有的计算研究60,61。
为了执行配体结合模拟,首先使用Omega Toolkit62生成MRTX1133的构象。然后,取最佳构象异构体,随机定向,并随机放置在蛋白质质心10Å半径内。然后使用MD Orion floes以与其他体系类似的方式对体系溶剂化。
为了使体系达到平衡,我们按照以下流程操作:首先,利用L-BFGS算法对体系能量进行最小化,收敛准则设定为2.4 kcal/mol。随后,在NVT系综中,以2.0 kcal/(mol·Å²)的力常数对KRAS、GDP以及任何共溶剂/探针分子的位置进行谐波约束,使其保持在初始位置,以便对体系进行10 ps的预热。最后,在NPT系综中,这些约束将在50 ps内逐步解除。在此平衡之后,系统在NPT系综中进一步模拟5 ns,以生成下游WE MD模拟所需的基础状态。所有NPT系综分子动力学(MD)模拟均基于OpenMM 7.7引擎63执行,采用随机(Langevin)积分器维持恒定温度与压力。其中,温度耦合设定为310 K,通过与热浴的弱耦合实现;压力控制则通过每0.5 ps与Monte Carlo气压计进行弱耦合,以维持系统在1 atm下的恒定压力。短程非键相互作用在10 Å截断半径处截断,长程静电相互作用采用平滑粒子网格Ewald(Smooth Particle Mesh Ewald, SPME)方法64进行高效处理。为实现2 fs的时间步长,系统中重原子与氢原子间的键长通过Constant Constraint Matrix Approximation(CCMA)65和SETTLE66算法进行刚性约束,从而有效提升积分效率并保持数值稳定性。
轨迹分析由OpenEye的OEChem67和Spruce54以及MDTraj68来共同完成。马尔可夫状态模型的构建则采用DeepTime69。
WE路径采样策略
分子动力学模拟的时间尺度受限于系统大小、力场表示、积分算法、MD软件的实现方式,以及用于模拟的硬件设备。在采用高度优化软件并基于传统硬件进行的MD模拟中,全原子力场的模拟时间可达微秒级别,这比KRAS蛋白中Switch-II口袋打开等生物罕见事件的速度快了数个数量级70,71。而WE方法则是一种无偏的稀有事件采样技术,能够有效跨越巨大的能量壁垒,并在极短时间内捕捉到超长的分子动力学时间尺度。该方法如同一个“统计棘轮”,沿着用户定义的“进展坐标”运行,实时追踪体系在任意物理可观测量上的演化过程25。
为增强KRAS构象状态的采样,我们并行运行了一组重复的分子动力学模拟(即“行走者”),每个独立的“行走者”都与其对应的概率(或“权重”)相绑定并实时跟踪。每轮分子动力学推进结束后,我们会执行一次重采样操作:对于进展坐标空间中占据不足区域的轨迹(称为“区间(bin)”),通过分裂方式增加其数量;而对于占据过多的“区间”,则通过合并来减少轨迹数量。其中,分裂是将一条轨迹复制并将其权重均匀分配给多个副本;而合并则是直接移除某条轨迹,并将其权重加到另一条幸存的轨迹上。经过多次动态推进与分裂、合并相结合的重采样迭代后,最终可生成一个包含大量加权轨迹的集合。在本研究中,我们为每个“行走者”在每轮迭代中持续推进了100皮秒的动力学过程。
针对表1中概述的每种模拟设置,均进行了500次WE重新采样迭代。由于WE算法在系统随时间演化过程中同时追踪各区间(bin)间行走者(walker)及其权重的转移,因此能够沿任何可能的反应变量精确估计动力学性质与加权种群分布(热力学性质)。WE分子动力学(WE MD)的一大优势是可在常规硬件上运行。通过合理选择进度坐标(progress coordinate)、行走者分配策略及分区间隔(binning procedure),该方法相较于暴力采样分子动力学(brute force MD)能显著降低稀有事件采样所需的计算成本。WE策略已在药物发现研究的多个其他领域中被证明是强大的机制研究技术,例如蛋白质-配体解离72、蛋白质折叠73与去折叠74、蛋白质-蛋白质结合75、类药物分子的被动渗透性76、SARS-CoV-2刺突蛋白激活77以及PROTAC三元复合物预测78。本文中,我们应用WE路径采样方法以探究KRASG12D蛋白中隐性口袋(cryptic pocket)开放的若干机制。
| KRAS Target | Cosolvent/Ligand(mM; no. per box) | ProgressCoordinates | Total simulation time (µs) | Wall clock time(Orion days) |
|---|---|---|---|---|
| WT | – | ANM mode 14 | 69 | 4.8 |
| ANM mode 15 | ||||
| xenon (150; 30) | ANM mode 14 | 40 | 2.8 | |
| ANM mode 15 | ||||
| G12D | – | ANM mode 14 | 70 | 3.5 |
| ANM mode 15 | ||||
| xenon (150; 30) | ANM mode 14 | 41 | 4.3 | |
| ANM mode 15 | ||||
| ethanol (150; 30) | ANM mode 14 | 40 | 3.5 | |
| ANM mode 15 | ||||
| benzene (150; 30) | ANM mode 14 | 42 | 7.3 | |
| ANM mode 15 | ||||
| MRTX1133 (5; 1) | ANM mode 14 | 45 | 9.0 | |
| Ligand RMSD | ||||
| ANM mode 12 | 48 | 6.4 | ||
| Ligand RMSD | ||||
| log of Ligand RMSD | 19 | 5.1 | ||
| Ligand distance | ||||
| Aggregate | 414 | 46.6 |
a每次模拟共运行500次迭代,其中最长的连续(非平衡态)轨迹长度可达50纳秒。
KRAS蛋白的正则模分析
本研究采用各向异性网络模型(Anisotropic Network Model, ANM)计算KRAS蛋白的正则模。首先,依照前述方法准备静态参比蛋白结构,选择了PDB ID: 4OBE中的KRASWT(野生型)结构作为参比。结构准备完成后,使用ProDy软件包以所有Cα原子(代表氨基酸残基的空间位置)构建ANM模型。通过该模型,可从Cα原子间相互作用的简谐势近似推导出的海森矩阵(Hessian matrix,维度为3N × 3N,N为残基数)计算得到一组正则模式。该海森矩阵的特征值(λ)对应于正则模式振动频率的平方。通常,矩阵存在六个零特征值,对应于体系的刚体平动和转动自由度。对于剩余的3N–6个分子内振动模式,其模式频率(ω)、振幅(A)及相应能量(E)之间的关系可表示为:
$$
\langle A^2 \rangle = \frac{E}{\omega^2}
$$
因此,给定单位输入能量,沿慢模式的位移往往比快模式具有更大的振幅。或者等效地,给定相同的振幅,沿慢模式的位移比快模式需要更少的能量。因此,特征值的倒数(λ−1),称为模式方差,通常用于表示沿给定模式变形的能量成本。分数方差(ρ)衡量给定模式解释的总方差的比例,定义为(对于第i个模式):
$$
\rho_i = \frac{\lambda_i^{-1}}{\text{tr}(H^{-1})}
$$
请注意,分母是(伪)逆Hessian的迹(trace),实际上是所有模式方差的总和。我们按相应的特征值对模式进行排序,并保留前20个(最慢模式)进行检查。Hessian的每个特征向量是模式形状,形式为(Δx1,Δy1,Δz1,···,ΔxN,ΔyN,ΔzN),其中Δxi,Δyi,Δzi是第i个Cα原子在给定模式中三个维度上的位移。由于特征向量是归一化的,Cα原子的位移大小是相对的。
虽然低频模式描述大振幅运动,但这种运动对于在整个蛋白质景观中搜索口袋可能仍然无效。例如,蛋白质的最慢模式可能局限于loop运动83,不一定是整个蛋白质表面隐秘口袋全局搜索的最有效运动。因此,需要一个指标来编码每个模式涉及的蛋白质量,这里我们使用模式集体性84作为该指标。模式集体性定义为:
$$
\kappa_i = \frac{1}{N} \exp \left\{ -\sum_{n=1}^N u_{i,n}^2 \log u_{i,n}^2 \right\}
$$
其中ui,n是第i个特征向量中第n个Cα原子的位移向量(Δxn,Δyn,Δzn)的范数。κ的取值范围在N−1和1之间,在最极端的情况下,当只有一个原子移动时κ=N−1,当所有原子在给定模式下以完全相同的振幅移动时k=1。直观地说,集体性表示参与沿给定模式运动的原子有效部分。从前20个最慢模式中,我们通常选择最具集体性的模式来指导蛋白质采样(见下文)。
KRAS的WE模拟中使用的进度坐标定义
正则模分析进度坐标(Normal Mode Analysis Progress Coordinate)
为了在WE模拟中真实地增强蛋白质运动的采样,可以构建一个进度坐标来测量沿蛋白质的一个或多个正则模式向量集合的演化。因此,对于每个WE迭代,使用MD传播动力学,然后计算当前构象相对于参考结构的变形向量δ。最后,将变形向量(δ)投影到参考模式(V)上,这实际上是两者的点积:
$$v = V^T\delta$$
其中V是作为列的特征向量矩阵。正则模式进度坐标v是当前模拟状态在选定正则模式上的投影。
配体RMSD进度坐标(Ligand RMSD Progress Coordinate)
定义了一个基于RMSD的进度坐标来跟踪MRTX1133结合到KRASG12D的相对位置和方向,该坐标使用PDB ID:7RPZ13的X射线晶体结构作为参考。为了执行RMSD计算,首先使用Spruce54通过叠加Cα原子来对齐参考和模拟蛋白质。将相同的平移向量和旋转矩阵应用于模拟中的MRTX1133,使其进入与蛋白质相同的参考框架。最后,使用OEChem67测量参考和模拟MRTX1133结构中重原子之间的原位RMSD。蛋白质对齐后MRTX1133的原位RMSD是本文中称为配体RMSD的量。
配体RMSD对数的进度坐标(Log of Ligand RMSD Progress Coordinate)
该坐标是前面讨论的配体RMSD进度坐标的简单对数变换,用于改善低配体RMSD值下结合MRTX1133构象的采样。
配体距离进度坐标(Ligand Distance Progress Coordinate)
另一个跟踪KRAS结合和未结合MRTX1133构象体之间相对距离的进度坐标以类似于上述配体RMSD进度坐标的方式计算。首先,使用Spruce54使用参考蛋白质(PDB ID:7RPZ)与模拟蛋白质对齐,并将相同的变换应用于模拟的MRTX1133副本。对齐后,使用OEChem67计算参考和模拟结构的原位质心(COM)位置。参考和模拟MRTX1133结构之间的COM距离是我们在本文中称为配体距离进度坐标的量。
SiteHopper相似性进度坐标(SiteHopper Similarity Progress Coordinate)
定义了一个基于SiteHopper的进度坐标,用于监测模拟框架中Switch-II口袋相对于MRTX1133结合的KRASG12DX-衍射结构中形成的口袋的形状和颜色(伪静电)相似性的组合。为了计算相似性,首先从MRTX1133在Switch-II口袋(PDB ID:7RPZ)内的KRASG12D结合位点定义参考SiteHopper补丁85。然后,对于WE模拟中的每一帧,使用排列MRTX1133结合口袋的残基定义一个单独的SiteHopper补丁。最后,使用OpenEye Spruce Toolkit54对模拟中的每一帧执行SiteHopper叠加,以最大化模拟框架和参考蛋白质结构的两个补丁的形状和颜色特征。7RPZ活性位点的结果相似性在本文中称为SiteHopper相似性,表示模拟Switch-II口袋相对于参考的3D形状和伪静电相似性。
KRAS的WE模拟
所有WE模拟都在平衡条件下进行,使用WESTPA2.0工具包86的Python API,该工具包已构建到Orion平台55的Small Molecule Discovery Suite中的一系列公开可用工作流中。使用最小自适应分箱方案(MAB)87在坐标空间的主体区域采样单维或多维进度坐标,在边缘附近使用单固定盒子以防止在感兴趣区域外进行浪费性采样。每个模拟进行了500次WE重采样迭代,对应于给定副本的最大连续(非平衡)轨迹为50 ns,这在Orion中使用约100个GPU平均需要约5天完成。从所有KRAS模拟生成的轨迹数据总量为414μs,这对于给定模拟根据使用的进度坐标的数量和物理性质而有所不同。所有WE模拟的详细描述,包括溶剂条件、使用的进度坐标以及总模拟时间列于表1。
从KRAS轨迹数据构建马尔可夫状态模型
对于暴露子和动态探针结合分析方法所需的互信息分析,我们从每个蛋白质模拟的WE轨迹构建马尔可夫状态模型(MSM),以生成其自由能景观的网络表示。首先,基于每个残基溶剂可及表面积(SASA)或残基-共溶剂距离向量的相似性对模拟采样的构象进行聚类。使用ZAP工具包88,89中可用的Gaussian OE Area方法计算每个残基的SASA。使用MDTraj计算蛋白质残基-共溶剂距离。在聚类构象后,计算每对聚类之间的转移概率,并通过每个构象的统计权重(即WE模拟权重)进行缩放。使用Deeptime69中实现的最大似然估计器从转移概率矩阵构建MSM。将聚类群体的平稳分布计算为转移概率矩阵的特征值为1的特征向量。在计算描述每对残基溶剂暴露或共溶剂结合相关变化的加权互信息时,使用MSM衍生的聚类群体。
暴露子(Exposon)分析
为了在KRAS中寻找口袋,我们使用了Bowman实验室开发的暴露子分析流程52。该分析从测量每对残基溶剂暴露的互信息(MI(X,Y))开始,使用以下方程:
$$
MI(X, Y) = \sum_{y \in Y} \sum_{x \in X} p(x, y) \log \left( \frac{p(x, y)}{p(x) p(y)} \right)
$$
这里,X和Y对应一对残基,x和y是它们的溶剂暴露状态,即埋藏或暴露,其中x和y的值对于埋藏为0,对于溶剂暴露为1。p(x)和p(y)是残基X和Y处于埋藏或暴露状态的概率。这些概率取自每个马尔可夫状态的MSM衍生群体。通过计算溶剂暴露的成对互信息构建残基-残基互信息矩阵。从这个矩阵中,使用亲和传播聚类算法90识别出在溶剂暴露变化中具有强相关性(高互信息值)的残基组,以及与剩余残基的较弱相关性。每个在溶剂暴露中具有高度协作变化的残基组称为暴露子52。
动态探针结合分析( Dynamic Probe Binding Analysis)
为了基于共溶剂结合行为的协作变化在KRAS中寻找口袋,我们假设这应该模拟诱导契合口袋开放的特征,我们使用氙与蛋白质中每个残基结合的互信息。这个想法受到上述e暴露子分析方法的启发,称为动态探针结合。在这种方法中,不是测量给定残基的溶剂暴露状态,而是使用氙(共溶剂)结合给定残基的概率来计算互信息。残基要么与氙结合,要么不结合,这种分类用于计算每对残基X和Y的结合互信息MI(X,Y)。残基X和Y处于结合/未结合状态的概率p(x)和p(y)从每个聚类的MSM衍生群体计算,如上述暴露子分析所述。使用亲和传播聚类算法90,将显示氙结合行为协作变化的残基组合在一起以寻找潜在的配体结合口袋。
探针图分析(Probe Map Analysis)
为了识别潜在的高亲和力隐秘口袋并提供自由能估计,进行了探针图分析。在这种分析方法中,计算氙原子在蛋白质表面相对于体水中的数量,这是口袋自由能的替代指标。为每个WE模拟框架在整个蛋白质-溶剂盒上生成3D网格。氙原子在1Å3网格单元内的占据率从基于残基-共溶剂距离向量相似性聚类轨迹数据获得的聚类中计算。网格占据率通过每个网格单元占据率与其所有面、顶点和边中心相邻单元(总共26个邻居)的占据率平均进行平滑处理。平滑的网格占据率(Ni)使用以下关系转换为自由能:
$$
\Delta G = -k_BT\ln\left(\frac{N_i}{N_0}\right)
$$
这里,N0是体溶剂中氙的预期网格占据率,设置为150 mM的均匀分布以模拟模拟设置。为了识别高亲和力结合位点,过滤掉自由能大于-2.5 kcal/mol的网格单元。剩余的高亲和力网格单元使用scikit-learn python包92中实现的DBSCAN聚类91方法进行聚类。对于DBSCAN聚类,两个网格单元被视为邻居的最大距离设置为3Å。
结果与讨论
使用WE MD沿正则模式作为进度坐标探索隐秘口袋开放
为了探索可能揭示口袋形成的KRASG12D结构动力学,我们进行了WE MD模拟,使用编码蛋白质构象景观信息的进度坐标。由于无偏、全面的隐秘口袋搜索需要探索感兴趣蛋白质的全局动力学,我们首先进行NMA以确定描述在平衡时发生最大结构波动方向的正交(orthogonal) 模式。根据能量均分定理,沿最低频率(最慢模式)的运动平均具有最大振幅。因此,这样的模式倾向于描述蛋白质的大规模波动93。此外,能量景观沿这些最慢模式的坡度最小,因此使用它们作为进度坐标来指导模拟将允许系统更容易地移出局部能量最小值,以探索更大但仍类似平衡的构象空间,从而有助于增强沿相关蛋白质运动的采样。
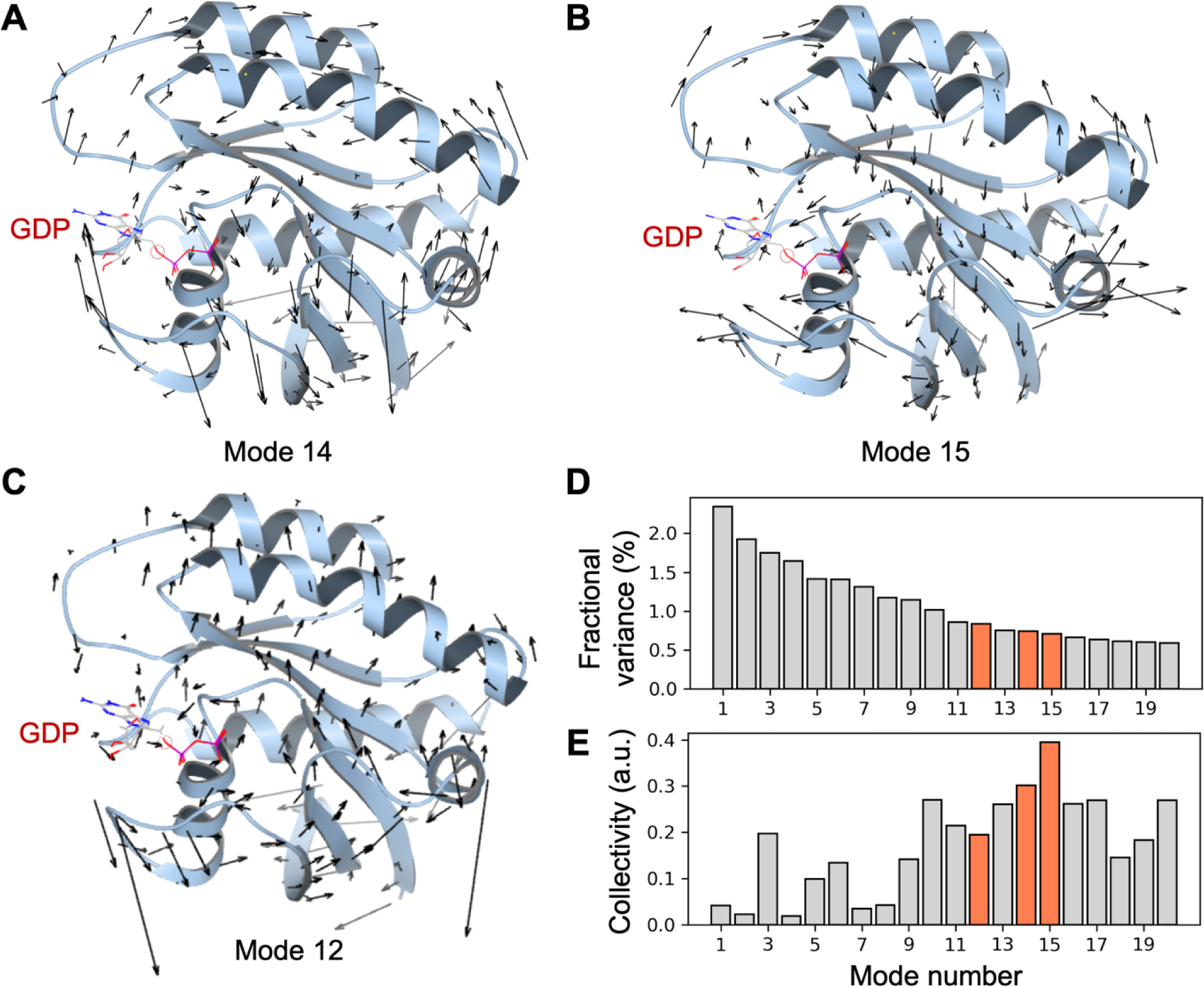 图2. KRASWT的正则模式析。基于野生型KRAS(PDB ID:4OBE)计算得到的(A)ANM第14模态,(B)ANM第15模态,(C)ANM第12模态。KRAS蛋白以浅蓝色飘带表示,黑色箭头指示从Cα原子位置(未显示)指向运动方向(切线方向)的模态形变矢量。(D)前20个最慢模态的模态分数方差。分数方差用于衡量某一特定模态所解释的整体动态占比(详见方法部分)。(E)前20个最慢模态的模态集体性。模态按(D)和(E)中分数方差大小降序排列。模态12、14与15以橙色高亮显示。
图2. KRASWT的正则模式析。基于野生型KRAS(PDB ID:4OBE)计算得到的(A)ANM第14模态,(B)ANM第15模态,(C)ANM第12模态。KRAS蛋白以浅蓝色飘带表示,黑色箭头指示从Cα原子位置(未显示)指向运动方向(切线方向)的模态形变矢量。(D)前20个最慢模态的模态分数方差。分数方差用于衡量某一特定模态所解释的整体动态占比(详见方法部分)。(E)前20个最慢模态的模态集体性。模态按(D)和(E)中分数方差大小降序排列。模态12、14与15以橙色高亮显示。
正则模式可以通过基于3D蛋白质结构的弹性网络模型(ENM)轻松估计,并包含固有分子运动的粗略表示79,80。ENM使用谐波振荡器网络来表示蛋白质复杂的分子内相互作用。ENM表示可以缩放到任何粗粒度,这里我们采用经典设置,其中网络中的每个节点(振荡器)代表一个蛋白质残基,每个边(弹簧)代表两个残基之间的相互作用。这种近似显著减少了描述蛋白质运动模式的参数数量,这些参数通常无法从需要原子力场的更详细模型中获得,然而ENM模型仍然产生与各种实验数据合理一致的结果29。然而,简单地使用最慢的ENM模式作为进度坐标来指导采样有一个注意事项。虽然最慢的ENM模式,即那些具有高分数方差的模式(图2D),通常表现出集体蛋白质行为,但它们可能被自由环、蛋白质末端甚至无序区域的运动所主导,这是网络模型采用的简单谐波近似的已知伪影。因此,为了识别描述集体蛋白质运动的慢而有意义的模式,我们还为每个模式计算了一个称为集体性84的度量,以定量评估特定模式对蛋白质结构的影响程度(图2E)。在这项工作中,我们采用了一种常用的ENM,称为ANM(见方法部分)来计算KRAS结构的正则模式。
为了以集体和有意义的方式指导蛋白质运动,我们使用最慢正则模式中前一个或两个最具集体性的模式的投影作为WE模拟中的进度坐标(见图2和表1;每个进度坐标上模拟的进展见图S3)。我们发现这种增强的蛋白质模拟程序是降低直接使用蛮力MD模拟运动所需的指数级大计算成本的便捷方法,而不会给Hamiltonian添加任何偏差。对于这项研究,我们选择了模式14和15用于KRAS中隐秘口袋的全局搜索。虽然这些模式仅描述了蛋白质整体动力学的很小一部分(\(<2%\))(图2D),但它们在指导隐秘口袋搜索的罕见事件采样方面是有效的(见下面的结果)。尽管如此,可以想象选择具有更大方差的较慢模式,但代价是模式集体性降低。
选择正则模式来指导WE模拟可能会影响某些蛋白质构象采样的效率,这反过来可能影响给定隐秘口袋在模拟过程中打开的能力。例如,如果局限于蛋白质特定区域的大规模构象变化对于隐秘口袋形成至关重要,那么选择在感兴趣区域具有相对低方差的最具集体性的模式可能会限制WE模拟中隐秘口袋形成的程度。在这种情况下,对于感兴趣残基具有高方差(由给定区域上的大模式向量振幅表示;见图2)的正则模式可能更有效地采样与涉及那些残基的特定隐秘口袋相关的动力学。慢模式的集体性也可以通过忽略蛋白质中较无序的区域来整体改善83。然而,自动确定蛋白质结构的哪个子域应该被突出显示或屏蔽以进行ENM构建以采样局部口袋(去)形成超出了本研究的范围。
从ENM衍生的正则模式的另一个限制是它们由于潜在的谐波假设仅在能量最小值附近准确。因此,其他使用正则模式作为偏置力的混合MD模拟方法需要在模拟期间不断松弛构象并重新计算正则模式(有关此类方法的综述,请参见参考文献94)。相比之下,在我们的WE模拟中没有对蛋白质分子施加外力,正则模式仅用作坐标来测量蛋白质构象从参考点沿模式向量指定的方向变形的程度。尽管如此,作为未来的方向,我们可以通过几种方式改进我们用作进度坐标的正则模式,以用于能量景观远处区域的构象。例如,我们可以:1)使用称为正则模式跟踪的技术95在非平稳位置跟踪和更新正则模式,或2)运行第二次模拟,使用基于第一次ENM模式模拟生成的构象通过基本动力学分析技术(如主成分分析(PCA)或时间滞后独立成分分析(tICA))解析的正则模式。
为了促进需要诱导契合机制开放的潜在隐秘口袋的识别,我们还在低浓度(150 mM)氙作为探针配体存在的情况下,使用上述正则模式坐标进行了WE模拟。
从加权系综轨迹中自动搜索潜在结合位点
为了检测可能由于构象波动而打开口袋的潜在位点,我们采用了三种不同的方法:(1) 暴露子分析,(2) 动态探针结合分析,和(3) 探针图分析,其中后两种分析方法需要使用(氙)共溶剂的模拟。
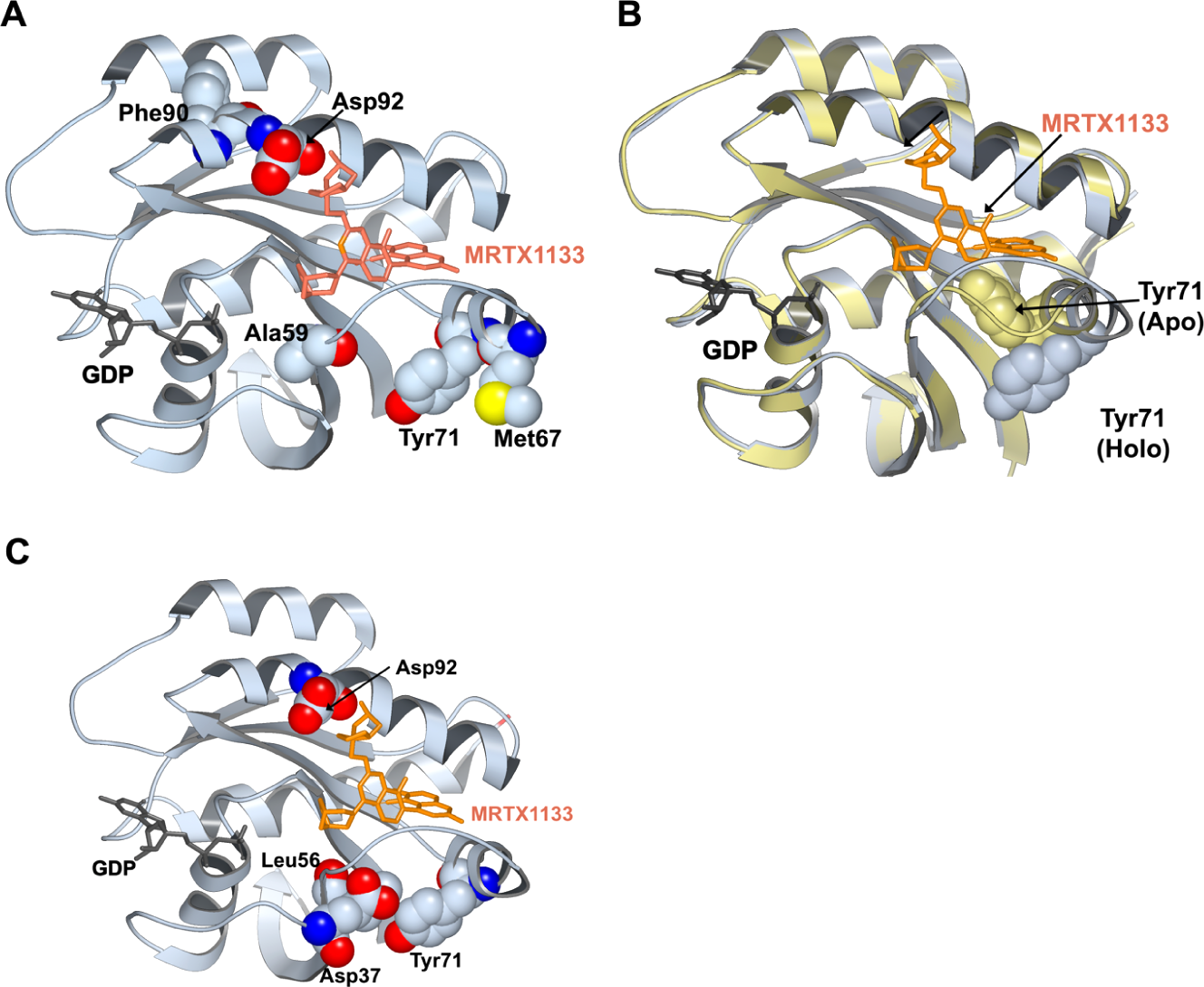
图3. KRASG12D的暴露子(exposon)分析与动态探针结合分析。(A)预测MRTX1133结合位点为潜在隐匿口袋的暴露子所包含的残基以球状形式展示,MRTX1133以橙色棍状模型表示。(B)无配体结合的KRASG12D:GDP复合物(浅蓝色)与结合MRTX1133的KRASG12D:GDP复合物(浅黄色)之间的结构重叠显示,在抑制剂(MRTX1133)结合状态下,Tyr71残基暴露于溶剂环境。(C)预测MRTX1133结合位点为潜在隐匿口袋的动态探针结合位点所涉及的残基以球状形式展示。
暴露子分析方法将潜在隐秘口袋识别为一组残基,这些残基由于蛋白质构象的波动而经历溶剂暴露的协作变化52。通过这些在溶剂暴露中的集体变化,暴露子残基预计将揭示潜在的结合位点。在这里,我们将暴露子方法应用于分析KRASG12D:GDP复合物的WE模拟,以查看是否有任何隐秘口袋会被预测(详见方法)。KRASG12D的暴露子分析揭示了几个可能可成药的位点。暴露子方法学识别的位点之一排列有形成Switch-II隐秘口袋的残基,MRTX1133和其他KRAS抑制剂结合在该位点(见图3A)。由于暴露子完全基于溶剂暴露的变化预测隐秘口袋开放,因此不经历相关变化的口袋形成残基不会被检测到。因此,一些不经历集体溶剂暴露变化的口袋排列残基,因为它们要么持续埋藏,要么持续溶剂暴露,不包括在预测(隐秘)Switch-II位点的暴露子中。
一般来说,暴露子分析非常适合寻找通过脱辅基蛋白的亚稳构象子集的构象选择存在的潜在可成药隐秘口袋。然而,如果诱导契合机制在口袋形成中起核心作用,这种方法很可能不会成功,特别是在没有类药片段或探针分子存在的情况下进行模拟时。暴露子分析的另一个限制是它不提供对检测位点可能结合亲和力的洞察,因为溶剂暴露的变化与口袋的潜在亲和力没有很强的相关性。在这项工作中,受到暴露子方法学的启发,我们提出了一种替代方法来克服这些限制,即在共溶剂存在下模拟生物分子。我们寻找隐秘位点的方法,称为动态探针结合,将潜在结合口袋定义为一组显示共溶剂结合行为协作变化的残基(见方法部分的理论背景)。在这项研究中,我们使用150 mM氙作为探针/共溶剂进行了混合溶剂模拟,以识别隐秘口袋。动态探针结合分析检测到多个潜在隐秘口袋,这些口袋通过构象选择或诱导契合机制打开。实际上,暴露子和动态探针结合分析方法预测了Switch-II口袋内已知MRTX-1133结合位点附近的互补残基(图3C)。有趣的是,我们发现Tyr71似乎根据口袋的关闭或开放状态分别采用向内或向外朝向的构象,这从b暴露子和动态探针结合方法都可以看出(图3B)。
暴露子和动态探针结合分析方法在概念上相似,因为它们都旨在通过测量蛋白质环境中的协作变化(通过它们的溶剂暴露或共溶剂结合行为)来识别隐秘口袋。因此,这些方法可用于寻找在MD模拟期间短暂开放的口袋。然而,在模拟期间快速打开并通过稳定结合探针分子保持开放的口袋可能无法被检测到。为了识别这种类型的口袋形成残基,可能包括高亲和力结合位点,我们使用探针图分析方法通过计算探针在3D网格上的结合自由能来分类隐秘口袋。这里,3D网格用于通过将网格占据率与体溶剂进行比较来找到稳定结合探针分子的蛋白质表面斑块。这种方法已与用作探针的有机共溶剂一起使用,以在几种蛋白质中寻找潜在药物结合口袋33,37,53,96。在这里,我们分析了使用WE路径采样策略进行的几个水-氙混合溶剂模拟,以寻找高占据率的氙结合位点。
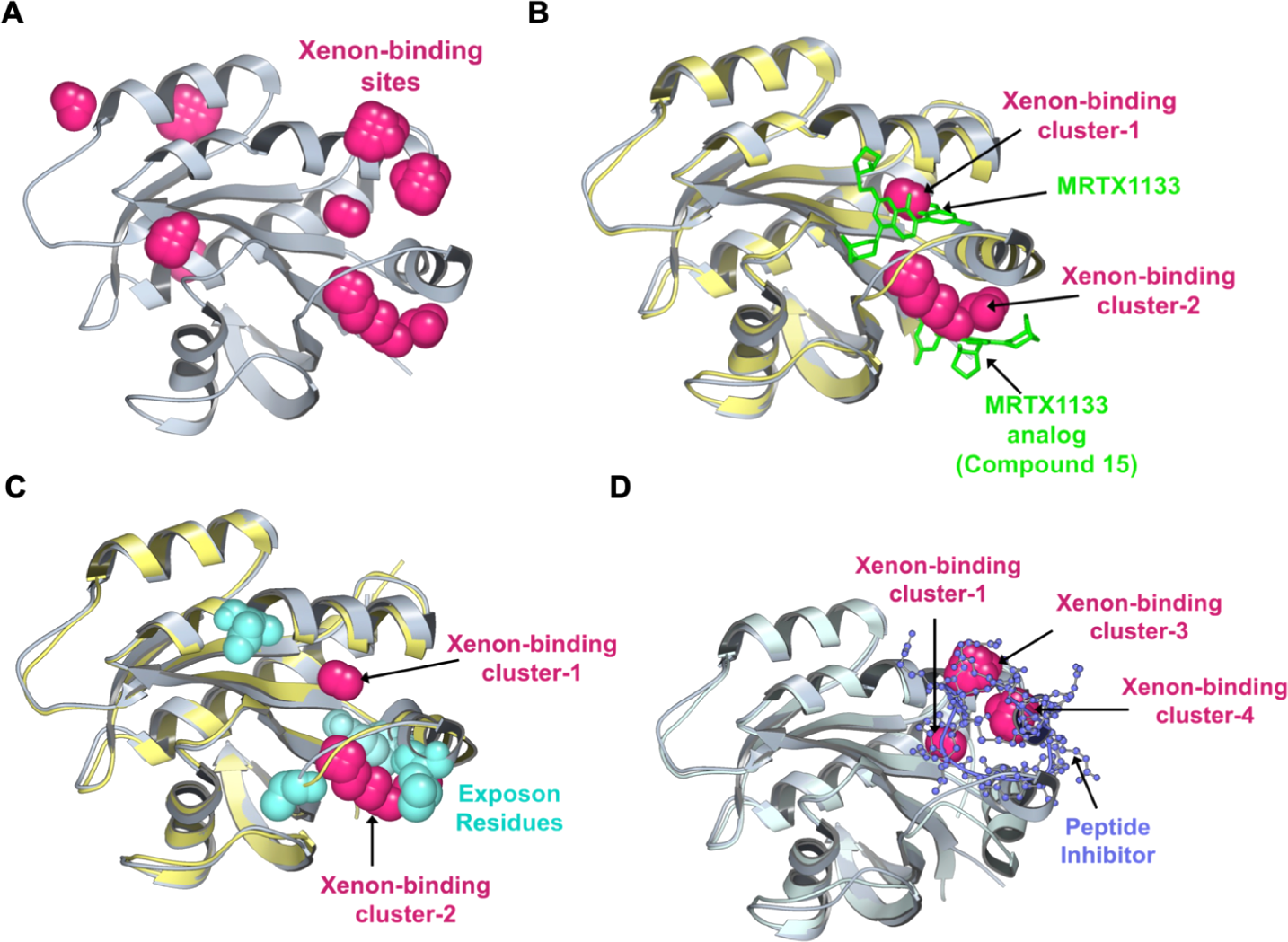
图4. KRASG12D的探针映射分析。(A)结合自由能低于−2.5 kcal/mol的氙(Xenon)结合格点以品红色球体表示。(B)氙结合簇-1与MRTX1133的结合位点重叠,而氙结合簇-2则与MRTX1133类似物的次级结合位点重叠。MRTX1133及其类似物以绿色棍状模型表示,其结合蛋白构象以浅黄色显示。(C)暴露子(exposon)所涉及的残基以青色球体展示;这些暴露子残基环绕着两个相邻的氙结合簇,且该区域与(B)中所示抑制剂结合位点高度重叠。(D)氙结合簇-1、-3和-4分别与一肽类抑制剂(紫色球棍模型)的结合位点发生重叠。在所有图中,模拟所得蛋白结构以灰色表示,参比结构则以浅黄色(PDB编号:7RPZ)或浅蓝色(PDB编号:5XCO)显示。
为了计算蛋白质表面上的氙占据率,我们首先基于其残基-氙距离向量的相似性对蛋白质构象进行聚类,并从这些聚类构建MSM以推导每个聚类的平衡群体。接下来,我们从放置在模拟盒上的网格中1Å3单元内氙原子的MSM加权占据率计算氙结合自由能。为了找到高占据率/亲和力的氙结合位点,过滤掉所有结合自由能大于-2.5 kcal/mol的网格点。这导致了8个高亲和力氙结合簇,可能包括潜在可成药的位点。其中,4个簇与已知的KRAS-抑制剂结合口袋重叠。氙结合簇-1与MRTX1133和Adagrasib的C7-萘基团重叠,两者都占据Switch-II结合位点中的一个深疏水子口袋(图4B)。值得注意的是,MRTX1133先导化合物在C7位置的官能团在优化活动期间被靶向以提高效力13。在簇-1附近的氙结合簇-2被发现与Switch-I/II口袋重叠,该口袋已在一系列晶体结构中被识别为结合小疏水分子15,16。事实上,MRTX1133的一个类似物(化合物15)也在晶体结构(PDB ID:7RT1)中发现占据这个口袋(图4B)。此外,在MRTX1133类似物化合物15的共晶体结构(PDB ID:7RT1,图4B)中,两个抑制剂副本结合,第一个在预期的Switch-II位点,第二个在次级Switch-I/II位点。KRAS中的这个位点也是与通过核苷酸交换激活KRAS的SOS1蛋白结合界面的一部分16。因此,人们可能想知道探针图分析是否也能预测可能的蛋白质-蛋白质相互作用界面。有趣的是,氙结合簇-1和-2也被上述暴露子残基包围,表明那些残基的溶剂暴露变化可能与两个相邻口袋的形成相关(图4C)。在簇-1附近的另一组氙结合簇被发现与KRASG12D-肽抑制剂复合物晶体结构(PDB ID:5XCO)97中观察到的肽抑制剂结合位点重叠。总的来说,通过使用氙作为探针,我们能够在KRAS突变体中定位三个已知的抑制剂结合位点。鉴于这些位点中的每一个都具有不同的残基组成并结合一系列不同的分子,氙似乎是在短时间内寻找隐秘(变构)位点的合理通用探针。虽然其他氙结合簇不与任何已知的抑制剂结合位点重合,但这些位点可能代表尚未探索的潜在可成药口袋。
诱导契合与构象选择机制在KRASG12D隐秘口袋形成中的作用
在使用暴露子和探针结合分析方法在KRAS中检测到Switch-II位点后,我们寻求深入了解这个隐藏了几十年的口袋的形成机制。应该注意的是,采样Switch-II口袋构象的能力完全取决于其形成机制。如果Switch-II口袋通过构象选择机制形成,只有能够由于精确的形状和静电互补性而容纳配体的蛋白质构象的小子集将存在于完整的蛋白质集合中98,99。在这种情况下,人们会期望开放构象的采样是限速步骤。相反,如果Switch-II口袋通过诱导契合机制形成,那么蛋白质-配体碰撞的生产力不依赖于蛋白质处于开放的蛋白质构象,这在没有配体的情况下不会被采样21,100。因为正确的配体总是会诱导与溶液中任何蛋白质的结合,蛋白质的诱导构象变化反而会限制kon。因此,人们可能通过精心设计的一组实验研究蛋白质-配体结合的动力学来解决机制细节。虽然在探针和配体分子存在下类全息结构的群体转移可以暗示配体结合机制,但监测配体结合和构象变化通量的动力学研究对于得出诱导契合机制和构象选择在不同条件下的贡献是必需的101−103。在这项工作中,我们分析了在探针或配体分子存在和不存在的情况下类全息构象的群体转移。
为了从计算角度了解Switch-II口袋的形成机制,即构象选择与诱导契合,我们对WT和G12D亚型的KRAS的所有WE模拟进行了SiteHopper分析,包括在纯水和几种共溶剂存在下的模拟。该分析将模拟中形成的Switch-II口袋构象的形状和静电与先前解析的KRASG12D与高亲和力小分子抑制剂(MRTX1133)结合的晶体结构进行比较。如果Switch-II口袋开放是由于蛋白质集合的构象选择,人们会期望在纯水中具有高SiteHopper相似性,而对任何探针分子的化学性质依赖性很小。然而,如果诱导契合机制主导Switch-II口袋开放,人们会期望在纯水中相似性低,随着探针分子趋向于MRTX1133,相似性逐渐增加。
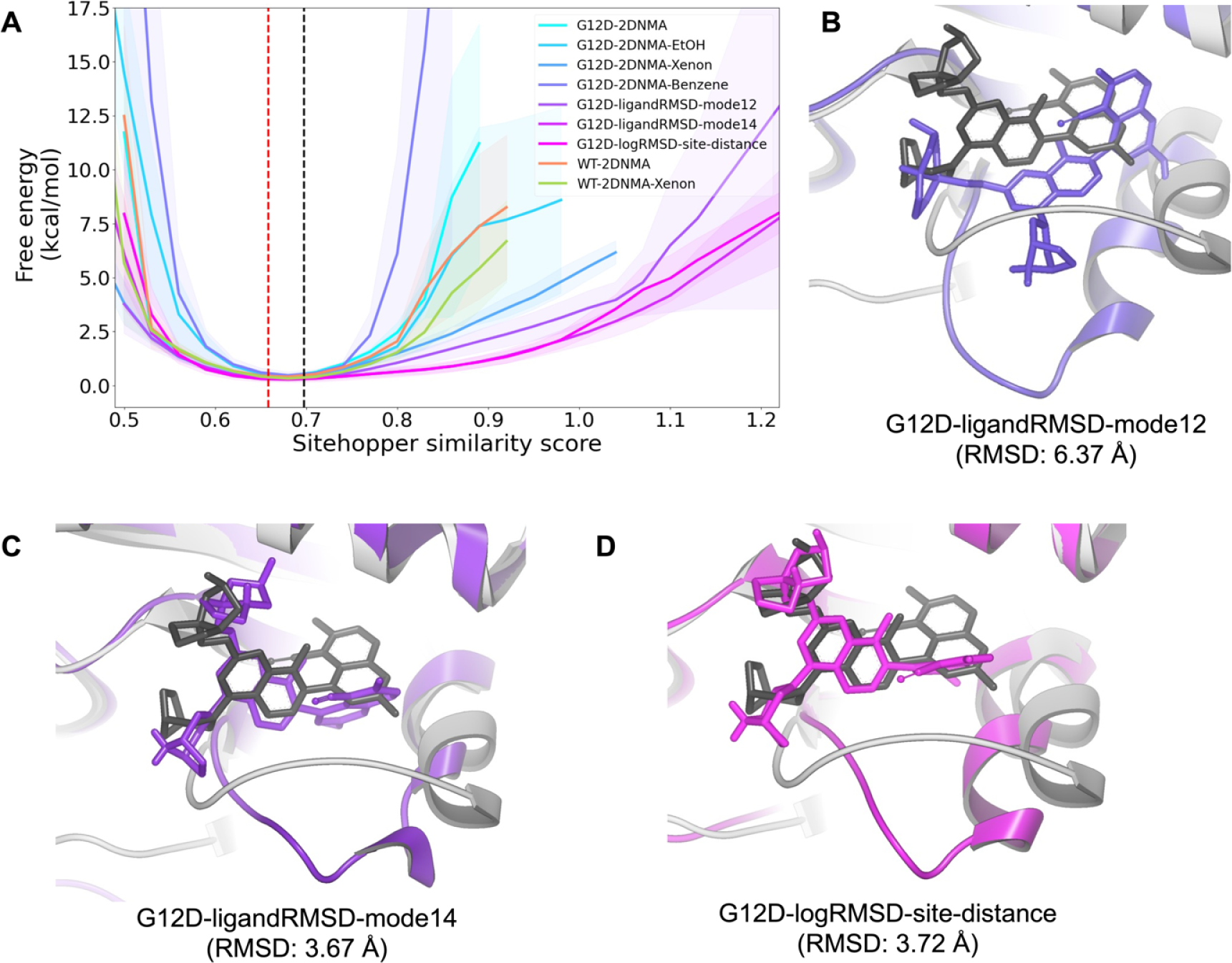
图5. KRAS WT与G12D突变型的SiteHopper相似性分析。(A)基于KRAS:GDP复合物在不同条件下的分子模拟计算所得的SiteHopper相似性评分的类自由能分布图。红色虚线对应于KRASG12D:GDP无配体结构(Apo构象)所计算的SiteHopper相似性评分;黑色虚线对应于用于启动加权平均分子动力学(WE MD)模拟的KRASG12D:GDP复合物平衡态结构所计算的SiteHopper相似性评分。自由能通过公式 \(-k_B T \ln[P(\text{SiteHopper相似性评分})]\) 计算获得。(B–D)晶体结构中MRTX1133结合模式(PDB编号:7RPZ)与各配体结合模拟中最低RMSD结合构象之间的重叠情况。晶体结构中的结合构象以灰色棍状模型表示。(B)在ligandRMSD-mode12模拟中观测到的最低RMSD结合构象以紫色显示;(C)在ligandRMSD-mode14模拟中观测到的最低RMSD结合构象以深紫色显示;(D)在logRMSD-site-distance模拟中观测到的最低RMSD结合构象以品红色显示。同时列出了各模拟中MRTX1133结合构象相对于晶体结构中结合模式的RMSD值。
在氙、乙醇、苯和MRTX1133作为探针分子存在的情况下,对KRAS的WT和G12D亚型进行了WE模拟。SiteHopper相似性分析显示,在共溶剂存在下的KRAS采用的Switch-II口袋构象比没有任何分子探针的模拟更类似于MRTX-1133结合结构(见图5A)。值得注意的是,与使用乙醇或苯作为探针分子的模拟相比,氙-水混合溶剂模拟中的SiteHopper相似性评分分布显示出更高的口袋相似性,以及更高概率(更低”自由能”)的相似性。然而,MRTX1133结合模拟获得高SiteHopper相似性评分的概率最高(”自由能”最低),并达到了最高的相似性值,这意味着与本研究中使用的其他化学探针相比,当MRTX1133存在时,Switch-II口袋更紧密地类似于晶体结构。总的来说,SiteHopper相似性分析表明诱导契合机制强烈促进了Switch-II口袋的形成,它也支持使用氙作为探针寻找潜在变构位点以及揭示需要诱导契合机制开放的可成药口袋的想法。
此外,使用两组不同的正则模式指导的MRTX1133结合模拟表明,可成药构象的采样并不显著受到作为WE进度坐标的正则模式选择的影响。如前一节所讨论的,沿ANM模式12采样将捕获排列结合口袋的Switch-II区域的慢动力学(图2)。人们可能预期在MRTX1133结合模拟中使用ANM模式12作为进度坐标比使用ANM模式14更能增强口袋形成。模式12更直接地捕获Switch-II口袋运动,而模式14更全局/集体。然而,我们对这些模拟的SiteHopper分析并没有显示在可成药构象分布上因选择正则模式作为进度坐标而有任何显著差异。这意味着体现蛋白质全局动力学的正则模式可能足以采样与隐秘口袋形成相关的局部动力学。
为了进一步了解与口袋形成相关的骨架和侧链采样的程度(图S1),我们还使用MRTX1133结合结构作为参考对口袋形成残基进行了RMSD分析。WE模拟的RMSD分布比较表明,在所有模拟中,包括在纯水中的模拟,结合位点残基采用类似于抑制剂结合晶体结构的骨架构象(即低RMSD)(图S1a)。相反,相对于抑制剂结合晶体结构计算的结合位点残基的全原子RMSD表明,只有在探针分子存在下,结合位点残基的可成药侧链取向的采样才会改善,当MRTX1133存在于模拟设置中时,侧链取向的改善最大(图S1b)。有趣的是,我们没有注意到MRTX1133结合结构的结合位点相似性与MRTX1133和结合位点之间的距离之间的强相关性。如果Switch-II口袋结合开放仅涉及诱导契合机制,人们会期望当MRTX1133接近结合残基时,结合位点残基采用更多的可成药构象。然而,我们对结合位点残基RMSD和MRTX1133与其口袋位置COM距离的相关分析没有显示这种相关性(图S2)。在我们使用各种进度坐标的MRTX1133结合模拟中采样的配体结合构象中,我们发现这些构象与晶体结构偏差超过3.7Å(图5B-D)。目前尚不清楚我们的MRTX1133结合模拟是否未能完全捕获完整的结合过程,或者构象选择机制在活性位点重新定向中起更大作用,而精确构象根本没有被采样。有趣的是,选择直接增强Switch-I/II区域(ANM12)采样的正则模式在找到正确的MRTX1133结合构象和活性位点骨架构象方面不如更具集体性的模式(ANM14)有效(图5B,C)。这一结果表明,全局蛋白质运动可能至少部分参与Switch-II口袋形成,并且可能也参与其他隐秘口袋形成,这强调了使用最具集体性的正则模式作为蛋白质采样的通用进度坐标的潜在效用。此外,使用配体RMSD的对数变换对于找到合理接近的MRTX1133结合构象(\(<4Å\))也很有用(见图5E)。考虑到找到正确蛋白质骨架和配体构象的问题,可能需要更长的模拟来获得关于MRTX1133与Switch-II结合的定量途径(动力学)和群体(热力学)信息。
结论
通过超过400μs的KRAS原子WE模拟,我们能够证明沿正则模式的投影可以是一种实用的进度坐标,用于在不知道连接状态集合的精确运动的情况下快速探索蛋白质构象景观。我们还提出了一种使用氙作为通用化学探针检测诱导契合隐秘口袋开放的方法。此外,我们展示了使用三种基于物理的方法从正则模式驱动的WE模拟产生的大量构象集合中自动检测潜在隐秘口袋。所有三种分析方法都回顾性地预测了实验研究报道的KRAS中的隐秘口袋。其中一个预测的隐秘位点,Switch-II口袋,结合了仅有的两种FDA批准的KRAS药物,Adagrasib和Sotorasib。本研究中用于隐秘口袋搜索的采样方法可以扩展到研究其他生物学和药理学相关的罕见事件。通过微调,这里提出的分析方法也可用于检测其他复合物中的隐秘位点,包括已成为现代治疗剂(如降解剂和分子胶)靶标的蛋白质-蛋白质或蛋白质-核酸界面。通过这种方式,正则模式可能被证明是通用生物分子动力学路径采样的有用进度坐标,特别是当靶标的期望(可成药)构象未知时。总的来说,这项工作引入了一个新框架,使用氙作为探针进行隐秘口袋搜索,并使用ENM衍生的正则模式作为有效的进度坐标,当隐秘口袋形成相关的动力学位置和性质几乎总是未知的情况下。我们未来的工作将致力于验证这一框架用于预测包括在蛋白质-蛋白质界面形成的隐秘口袋在内的多种蛋白质中的隐秘口袋。
数据与软件
已提供补充数据以重现本研究,包括GDP结合的野生型和突变型KRAS蛋白的初始结构(PDB格式)以及所有化学探针的结构(SMILES字符串)。WE MD模拟是使用WESTPA工具包和OpenMMMD引擎作为动力学传播器进行的;这两个工具包都是公开可获取的。化学探针的小分子构象生成是使用商业软件的OpenEye Omega Toolkit完成的。包括G12D突变在内的初始蛋白质结构准备是使用商业软件OpenEye Spruce Toolkit进行的,而初始分子动力学系统构建是使用免费可用的PACKMOL完成的。蛋白质力场参数取自公开可用的Amber14SB力场。小有机分子的力场参数取自公开可用的OpenFF-2.0.0 (Sage)。氙的力场参数取自Warr等人发表的研究(60)。分子动力学模拟轨迹的分析是使用商业软件OpenEye OEChem和Zap Toolkit进行的。所有OpenEye Toolkits(包括本研究中使用的Spruce、Omega、OEChem和Zap Toolkit)也提供免费的学术许可。由于轨迹文件巨大,本研究的所有模拟和分析数据都存储在Orion上,可根据请求共享。在本研究中用来可自动化模拟和分析的OpenEye隐秘口袋检测工作流(OpenEye Cryptic Pocket Detection Floes)可在Orion的学术版堆栈上获得,也可向我们索取。
关于OpenEye隐秘口袋识别工作流(Cryptic Pocket Detection Floes)
对于某些靶标蛋白质,现有的静态结构可能缺乏明显的小分子结合位点,或者可见的口袋可能具有使其对药物开发具有挑战性的特性(例如,高家族相似性)。OpenEye隐秘口袋检测流(Cryptic Pocket Detection Floes)旨在揭示隐藏的结合位点,并帮助评估这些靶点的成药性。工作流通过使用加权系综分子动力学(MD)模拟来采样,然后分析产生的轨迹数据以识别潜在的隐秘口袋,其他OpenEye药物发现工具可在此基础进行后续的研究与探索。
更多关于OpenEye隐秘口袋识别工作流请访问:Cryptic Pocket Detection Floes
视频回放 | 攻克不可成药靶点:隐秘口袋检测与排序的高精度方法
主讲人

David LeBard, Head of Science at OpenEye
David在复杂生物系统的理论和计算建模方面均有背景,致力于促进工业界和学术界的合作,以寻找药物发现问题的解决方案,包括类药分子的被动渗透性建模、生物分子的稀有事件采样以及蛋白质中隐秘口袋的检测。
内容简介
为拓展可成药蛋白质组范围(涵盖KRAS等难成药靶点),我们开发了一套自动化工作流,使非专业人员也能评估蛋白质的配体结合能力。某些危及生命的疾病相关蛋白难以治疗,究其根本是缺乏明确的分子抑制剂结合口袋。典型代表KRAS蛋白——一种参与超过25%人类癌症的GTP酶。尽管学术界与工业界投入巨大努力寻找其可用口袋,但KRAS在过去三十余年中始终被视为不可成药靶点。
仅需蛋白质结构数据(X射线晶体、冷冻电镜、AI预测等),本工作流即可通过加权集成路径采样方法捕获稀有蛋白质构象状态,并运用马尔可夫状态模型组协同分析形成口袋的关键残基。进一步地,我们采用神经网络模型评估口袋内配体结合潜力,据此对预测口袋进行可药性排序。该流程能识别通过两种机制形成的口袋:稀有蛋白质构象选择或探针分子诱导契合。
本此视频会议中,以KRASG12D为概念验证案例,证明该方法可准确预测已知隐秘口袋(包括隐匿数十年的Switch-II口袋)。最后,我们将展示该自动化蛋白质采样与隐秘口袋检测流程在19种蛋白质(涵盖24类独特口袋类型)中的验证结果。
如何获取OpenEye Cryptic Pocket Detection Floes
OpenEye隐秘口袋识别工作流可从OpenEye云计算平台ORION获取,由于美国出口政策的原因,该ORION云计算平台对大中华地区不开放,只能以计算服务(FTE)的方式来使用。
常见问题
问:在进行隐秘口袋识别工作流计算时如何选择起始的结构?
答:运行隐秘口袋识别工作流时,起始结构的选择取决于具体的目标。若用户旨在验证工作流的可靠性,起始结构应选用目标蛋白的无配体(apo)构象。若用户针对特定靶标蛋白开展隐秘口袋识别,则应选择最能代表靶标生物状态的蛋白构象作为起始结构。例如,若旨在发现与蛋白质寡聚态相关的隐秘口袋,必须确保起始结构处于正确的寡聚状态。同样地,若需在特定底物结合状态下识别隐秘口袋,则应选择相应的底物结合态作为起始结构。以KRAS蛋白为例,若要识别其非活性状态下的隐秘口袋,起始结构必须在核苷酸结合位点存在GDP分子。


