
摘要:本文以DDR1抑制剂Ponatinib为起始化合物,用SPARK对其进行了基团替换计算实验。结果表明,SPARK在不到10分钟的时间内完成了计算,不仅获得了Alex等人采用深度学习生成化合物1的甲基哌嗪衍生物,而且该类化合物被SPARK打分后排序靠前(排名第一)。这说明SPARK比起深度学习在使用上可以更加简单、直接地获得目标化合物。
肖高铿/2019-09-05
深度学习生成DDR1抑制剂
英科智能(Insilico Medicine)Zhavoronkov Alex等人最近在Nature Biotechnology杂志上发表关于深度学习加速DDR1激酶抑制剂发现的文章1。药明康德微信公众号文章《从靶点到候选分子仅需3周!》2报道并总结了该文的亮点, 并评论说:“值得一提的是,在人工智能技术与研发人员的协同下,在选定靶点的46天后,新筛选出的分子就完成了初步的生物学验证”。科技日报9月3日撰文《人工智能发掘潜在新药仅需四十六天》3进行了报道:“尽管AI系统设计的药物似乎并不比传统研究方法开发的DDR1抑制剂更有效,但与开发候选药物的传统方法需要8年多时间和数千万美元的开发费用相比,因斯里克公司(即Insilico Medicine)的新方法仅需数周时间,成本大约15万美元”。
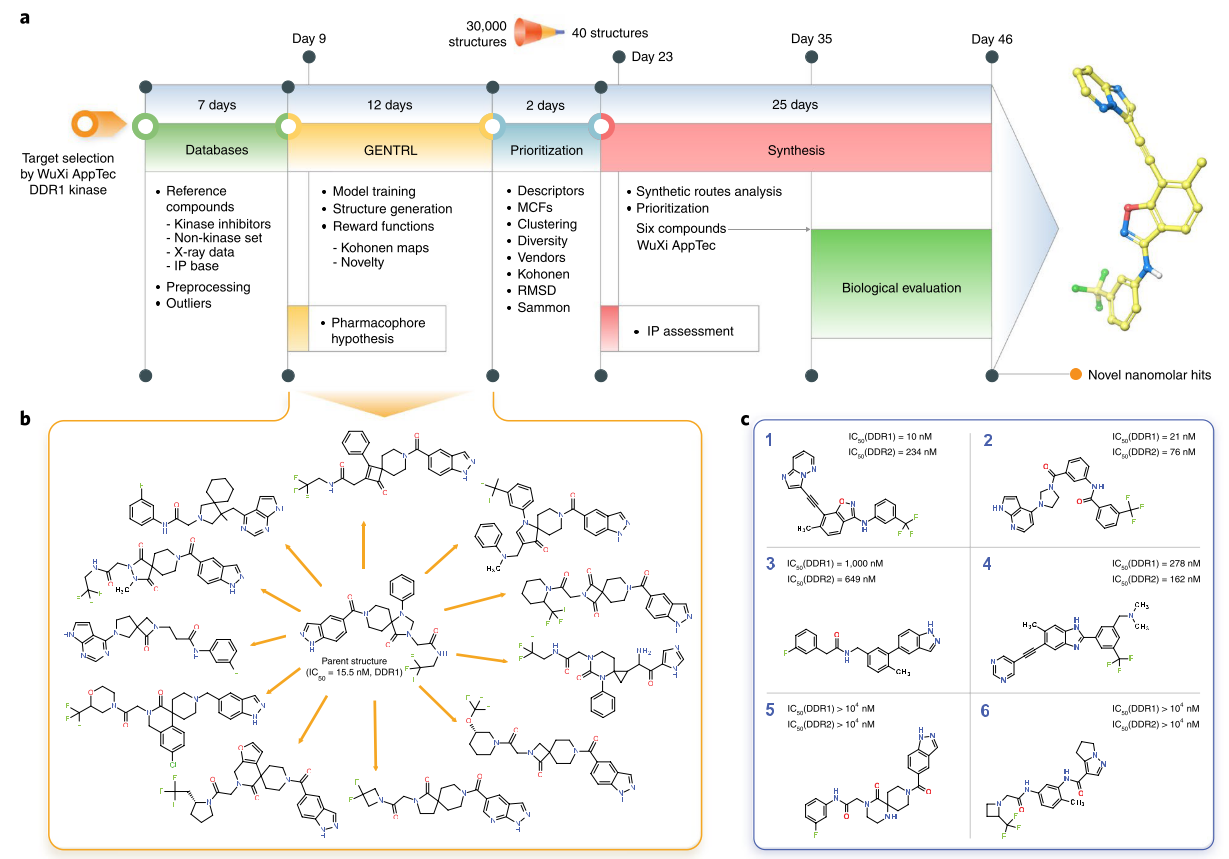
Figure 1. GeNTRL模型的设计、工作流与nM苗头化合物。a,GeNTRL分子设计总的工作流与时间线. b, 已知的DDR1激酶抑制剂与生成的代表性结构;c, 生成的对人类DDR1激酶抑制活性最强的化合物
Figure 1总结了抑制剂发现的流程与时间线;展示了生成的代表性化合物与实验验证的活性最强DDR1抑制剂。总的来说,深度学习确切无疑地加速了药物的发现过程,经过实验测试与验证,化合物1(Figure 2)被认定为最有发展潜力的候选化合物(原文为Lead candidate,我把它理解为candidate)。
Figure 2. Alex等人1用深度学习设计的化合物1
鉴于Alex的化合物1与已知的DDR1抑制剂Ponatinib(Figure 3)在结构上有很深的“渊源”, 有经验的药物化学家认为:化合物1的1,2-苯并异噁唑片段(Figure 2的黄色高亮部分)是Ponatinib苯甲酰基(Figure 3的高亮部分)显而易见的生物等排体,所以化合物1事实上是Ponatinib的me-too开发。视频1演示了基于结构的Me-too计算实验:通过观察Ponatinib与DDR1的相互作用,可以发现C=O作为氢键受体与蛋白ASP784发生氢键作用,将该C=O氧用SP2的N氮代替并环合,可以保留该相互作用同时保留分子的形状不变。
Figure 3. 已知的DDR1抑制剂Ponatinib,黄色高亮部分被生物等排体替换可获得化合物1的衍生物
视频1:利用化学经验对Ponatinib进行基于结构的me-too计算实验: 通过观察Ponatinib与DDR1的相互作用,可以发现C=O作为氢键受体与蛋白ASP784发生氢键作用,将该C=O氧用SP2的N氮代替并环合,可以保留该相互作用同时保留分子的形状不变。
我感兴趣的是:(1)用传统药物设计技术,比如基团替换策略能否重现Alex等人1的结果?(2)需要多长时间?(3)设计出来的化合物能否排序靠前以便药物化学家们马上关注到该化合物?为此,用SPARK的生物等排体替换策略尝试对DDR1抑制剂Ponatinib进行基团替换,以Alex等人的化合物1为目标考察计算结果,并与大家分享结果。
用SPARK对DDR1抑制剂Ponatinib进行生物等排体替换
从PDB下载Ponatinib与DDR1激酶的复合物晶体结构(3ZOS),并将Ponatinib作为SPARK基团替换的起始化合物(Starter),基团替换时仅搜索ChEMBL的常见片段库对Ponatinib的苯甲酰基片段(Figure 3黄色高亮部分)进行替换,具体过程见下面视频。
视频2:用SPARK对Ponatinib进行基团替换的实验: SPARK生成了化合物1的甲基哌嗪衍生物以及其它的备选化合物
如视频2所示,我们仅用ChEMBL(Version 23)的115610个常见片段进行了筛选(见Figure 4),计算采用默认值,最大输出500个打分最佳的化合物。整个过程10分钟不到就完成计算、获得计算结果。
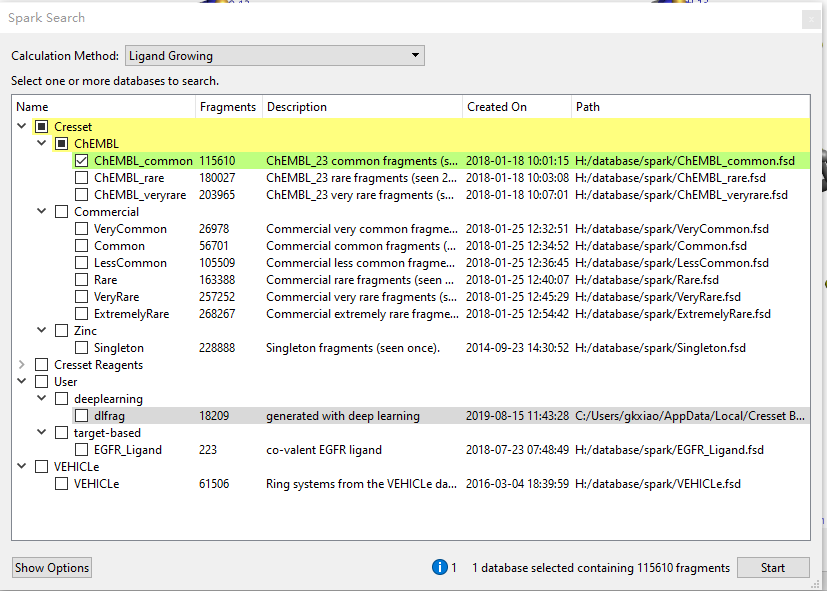
Figure 4. 基团替换仅对ChEMBL(Version 23)的115610个片段进行筛选
Figure 5所示展示了打分最高的前48个化合物(仅展示替换片段),您会发现Alex等人1化合物1骨架排序第1。
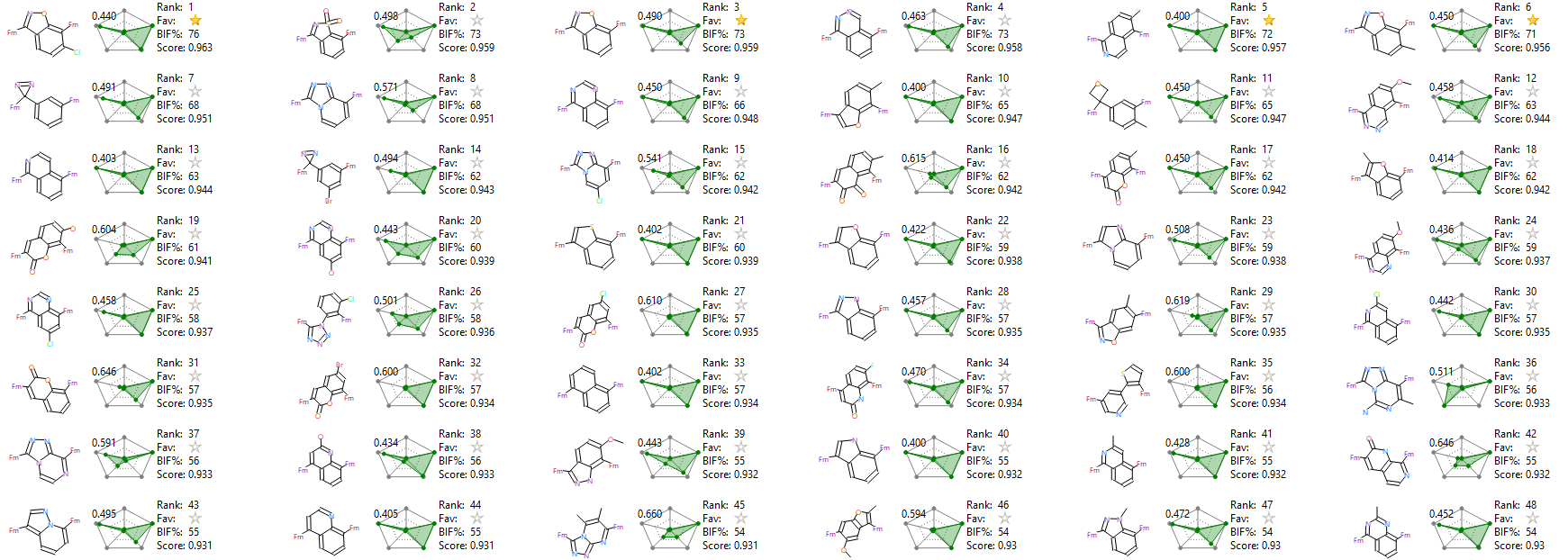
Figure 5. 打分最高的前48个化合物(点击图片切换至大图片)
对500个结构进行聚类,共得到214中不同的结构类型,按打分从高到低排序,其中排序第1的类别含有三个化合物正是Alex等人深度学习生成的化合物1,如Figure 6所示。Figure 6也给出了这三个化合物的骨架来源,分别来自CHEMBL523516,CHEMBL3338562与CHEMBL493496。
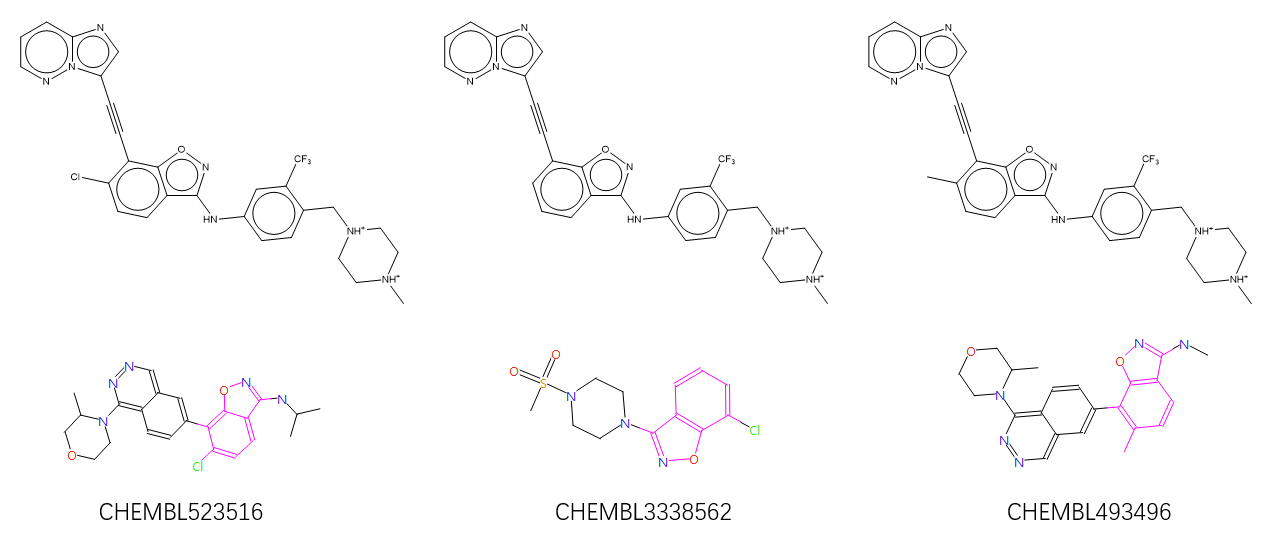
Figure 6. 打分最高的Cluster 1包含三个化合物(上)及其CHEMBL骨架来源(下),三个化合物在全部500个化合物排序分别为1、3与6(从左到右)
Figure 6右边化合物是Alex等人的化合物1的甲基哌嗪衍生物,SPARK的基团替换策略重现了Alex等人的深度学习从头设计结果;除此之外,还生成了羰基对位去甲基或氯取代的衍生物,该类化合物被SPARK的形状与场点相似性综合打分排序第一,药物化学家可以马上关注到该类化合物;虽然也需要对SPARK的结果进行谨慎地选择与后处理,但是在这个练习中不需要进行层层地性质过滤、药效团过滤等等操作就富集到了目标化合物。
视频3:Ponatinib及基团替换后化合物与DDR1的相互作用模式
结论
本文以DDR1抑制剂Ponatinib为起始化合物,用SPARK对其进行了基团替换计算实验。结果表明,SPARK在不到10分钟的时间内完成了计算,不仅获得了Alex等人采用深度学习生成化合物1的甲基哌嗪衍生物,而且该类化合物被SPARK打分后排序靠前(排名第1)。整个过程没有对化合物进行各种过滤,这说明SPARK比起深度学习结构生成在使用上可以更加简单、直接地获得目标化合物。
文献
(1) Zhavoronkov, A.; Ivanenkov, Y. A.; Aliper, A.; Veselov, M. S.; Aladinskiy, V. A.; Aladinskaya, A. V; Terentiev, V. A.; Polykovskiy, D. A.; Kuznetsov, M. D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019. https://doi.org/10.1038/s41587-019-0224-x.
(2)药明康德. “《自然》子刊:从靶点到候选分子仅需3周!药明康德协助研究”. 药明康德微信公众号(Wechat ID:WuXiAppTecChina),2019年9月3日. 2019年9月5日访问
(3)冯卫东. “人工智能发掘潜在新药仅需四十六天”. 科技日报, 2019年9月3日. 网址:http://digitalpaper.stdaily.com/http_www.kjrb.com/kjrb/html/2019-09/05/content_430066.htm (accessed Oct 9, 2019).


