
摘要:本研究将密度泛函理论(DFT)计算与电化学阻抗谱(EIS)测试相结合,提出了镍团簇催化次磷酸氧化还原的新机理。DFT计算结果表明,OH-的浓度是控制次磷酸氧化与还原的关键因素。高浓度的OH-有利于次磷酸的氧化反应,因为OH-可以直接与H3PO2)和氢自由基(Hc)结合。相反,在这种情况下还原反应被抑制,因为氢自由基优选与OH-而不是H3PO2结合。因此,pH值是控制偶联反应途径的关键开关。EIS结果表明,化学镀镍过程包括三个电化学过程:双电层的充放电,Ni(I)转变为Ni(II)或Ni(0)以及中间产物的特异性吸附。实验测试与理论预测吻合良好,实验测量结果表明:在低pH值下成功合成了具有高磷含量的化学镀镍涂层,该涂层具有非磁性,并且可以用作硬盘驱动器基板的非磁性涂层。
编译:孙鹏/中山大学
日期:2020-03-30
原文:Cui, G.; Liu, S.; Wang, K.; Li, Q.; Wu, G. Discovering P-Doped Mechanism in Non-Magnetic Ni–P Films for HDD Substrate: A Combined Experimental and Theoretical Study. RSC Adv. 2014, 4 (28), 14663–14672. https://doi.org/10.1039/C3RA47217E.
前言
由于高P含量(大于20%)的Ni-P化合物无磁性化,化学镀Ni(EN)工艺已经广泛应用于硬盘驱动器(HDD)基底。因此,如何有效地在EN过程中控制P的含量尤为重要。EN过程是复杂的多相催化过程。其中,P的掺杂是由于过程中加入的次磷酸还原剂。在沉积Ni的过程中,次磷酸的氧化和还原过程会同时发生。其氧化会产生亚磷酸盐,而还原会生成P,从而导致EN中P的掺杂。然而,目前为止,其氧化还原对的机理仍未完全理解,主要是由于共用的次磷酸反应物的耦合。
次磷酸的氧化机理有两种可能的反应轨道,其中一种由Van den Meerakker提出,认为次磷酸直接释放出一个氢自由基(H•),另外一种路径由Homma提出,认为次磷酸应该先与氢氧根相结合。此外,Homma对所有可能的中间体的相对能量进行了理论计算(DFT, MP2/6-311G(d,p)), 获得反应能垒为423.4 KJ mol-1。然而,在这些计算中,过渡态及Ni相关的催化行为并未考虑。相比之下,Homma提出的反应模型更加有可能发生。因此,再次我们主要集中研究Homma提出的反应路径,对其进一步阐释和完善。
对于次磷酸的还原机理,也有两种可能的反应机理。其中一个路径是Brenner提出的直接产磷机制。他认为次磷酸首先失去两个氢原子,然后是两个P-O键的断裂。另一种路径是由Saitou提出的间接产磷机制,其中PH3被认为是关键的中间产物。在这个机理中,H3PO2首先断裂两个P-O键,然后一个氢自由基与PH2中的一个磷原子结合形成PH3。镍磷合金电镀过程中的电化学测量数据证实了这两种可能的机理。在这项工作中,我们将在不考虑外部电场的情况下分析以上双种反应机制的可行性。
许多研究人员试图通过电化学或量子化学建模方法来研究其机理。然而,结果表明,单一的方法不能完全阐明该耦合过程。在这项工作中,我们通过电化学和量子化学建模相结合的方法来探索双反应机理。研究结果为如何控制次磷酸氧化还原反应的路径,准确预测磷矿的磷含量提供了新的思路。
此外,为了了解偶联反应的路径,我们还研究了OH–的作用。在EN过程中,发现不同浓度的OH–能够调节次磷酸酯的还原反应,并首次提出了这种耦合反应的OH–转换机制。实验结果表明,DFT法和ElS法的结合是分析这一复杂电化学过程机理的有效方法。
结果与讨论
磷含量对EN膜磁性能的影响
不同pH溶液对EN沉积物中磷的含量如图1所示。当pH值从5.0增加到9.0时,含磷量由13.8% wt%逐渐降至4.3wt%。当pH值等于1.0时,相应的磷质量含量下降约2.0%。
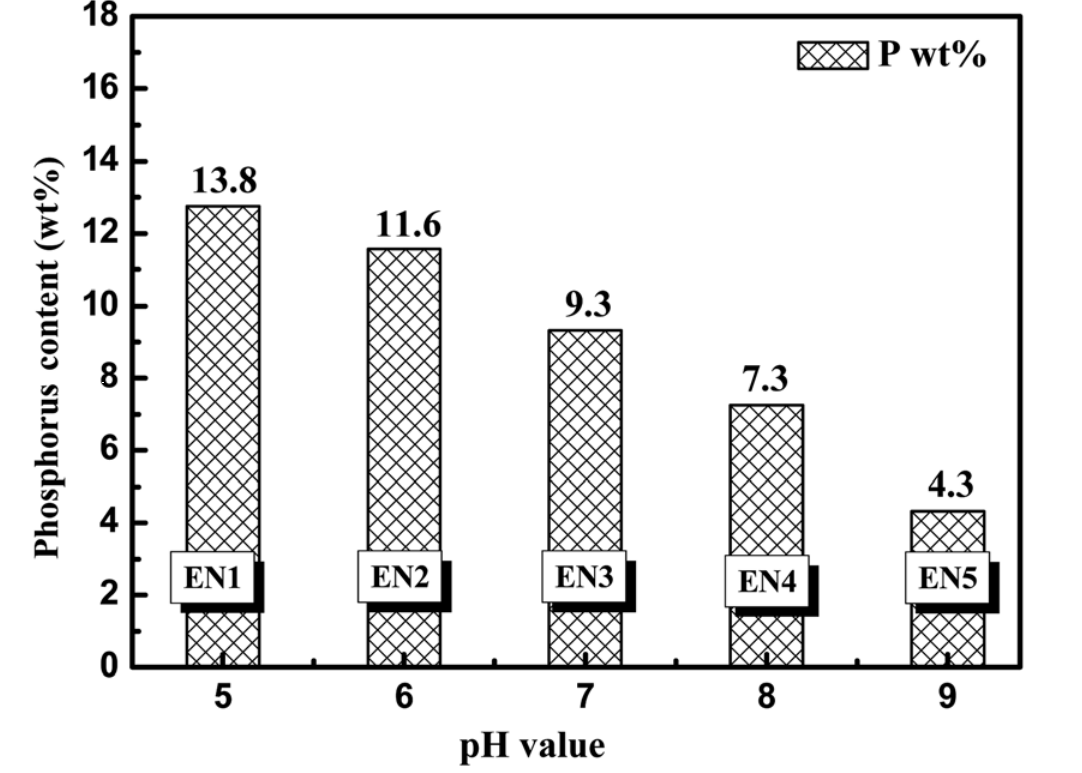
图1. EN沉积Ni-P涂层中磷含量(由XRF测定)与溶液pH值的关系(pH值范围为5.0到9.0)。
随后,测定了EN沉积物的磁性随EN样品中磷含量的变化(图2)。显然,与图2a中的其他EN样品相比,EN5具有磁性。磁力线的斜率从EN4逐渐减小到EN1,如图2b所示。此外,由于磷界面磁畴的存在,随着磷含量的增加,磁畴面积也减小。此外,随着掺磷量的增加,EN膜的结构由晶态向非晶态转变。一个非磁性EN在不干扰磁性记录材料的情况下,作为HDDs的基片时,涂层是必不可少的。因此,EN1具有较高磷含量的薄膜,更适合作为HDD衬底。
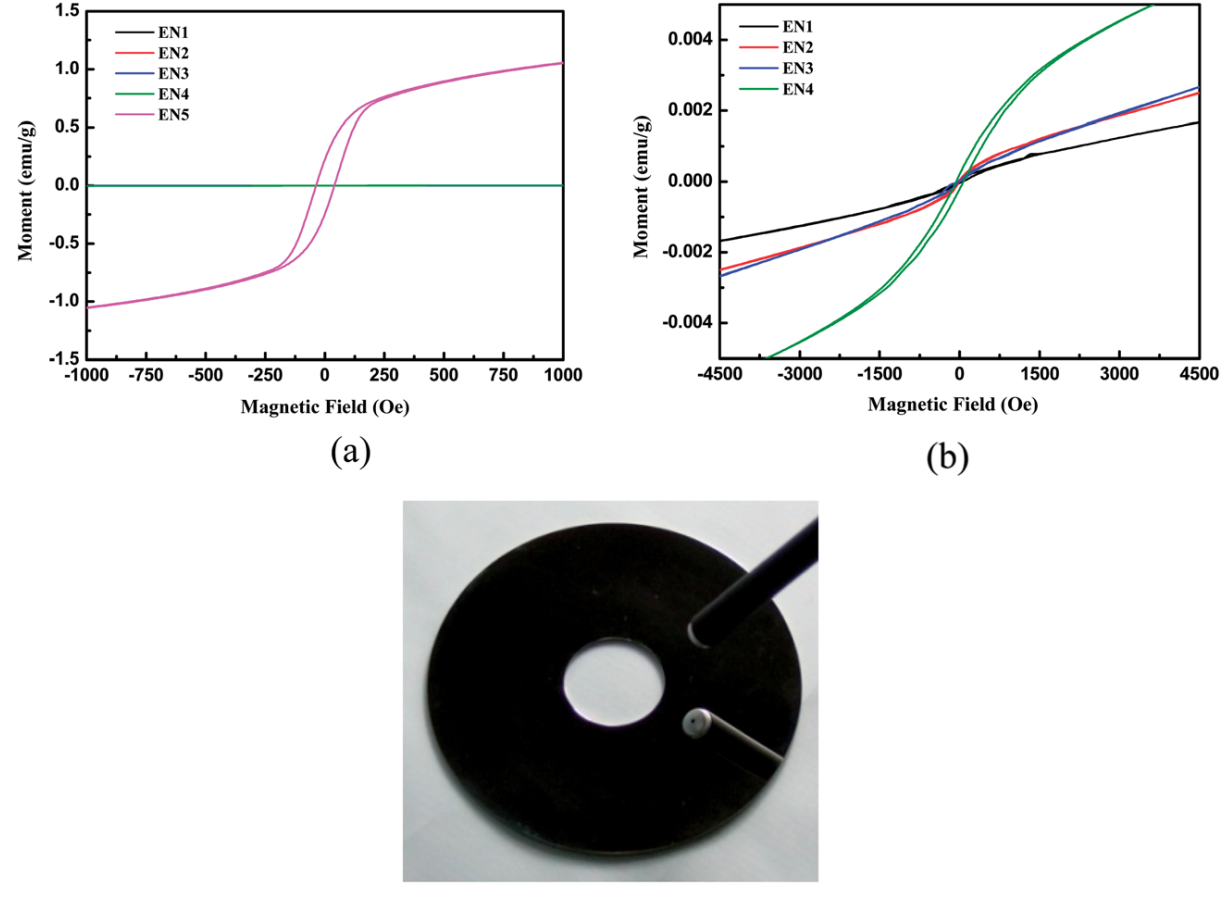
图2. EN沉积的Ni-P薄膜(a和b)的磁性能随磷含量的变化,以及在EN1条件下得到的HDD磁盘图像(c)。
来自次磷酸的耦合反应路径
次磷酸与Ni(II)化合物的耦合反应路径如示意图1所示。反应路径包括一个氧化路径(OP)和三个可能的还原路径(DRP-I、DRP-II和IRP)。在OP路径中,[Ni-(H3PO2)]2+首先与OH–反应形成IM1。随后,[Ni-(H4PO3)]+分解并释放一个氢自由基和一个电子。该反应路径最早由Homma提出,对于DRP-I路径,[Ni-(H3PO2)]2+依次释放2个H•。随后,P=O双键通过氧原子的加氢变成单键。最后。[Ni-(H3PO2)]2+ (IM3)失去两个OH–。在DRP-II路径中,从IM1到IM3过程中发生了一个氢分子间的转移过程,将其与DRP-I路径区分开来。首先,[Ni-(H3PO2)]2+释放一个氢自由基。接下来,P=O双键通过氢分子间转移从磷原子转移到氧原子而断裂。随后,[Ni-(H3PO2)]2+释放两个OH–,形成一个吸附P原子。在IRP路径中,P-O键比P-H键优先断裂。[Ni-(H3PO2)]2+(R)转化为[Ni-(PH2)]2+,同时失去两个OH–。随后,两个氢原子被释放为氢自由基。最终,P原子掺杂进入Ni镀层。
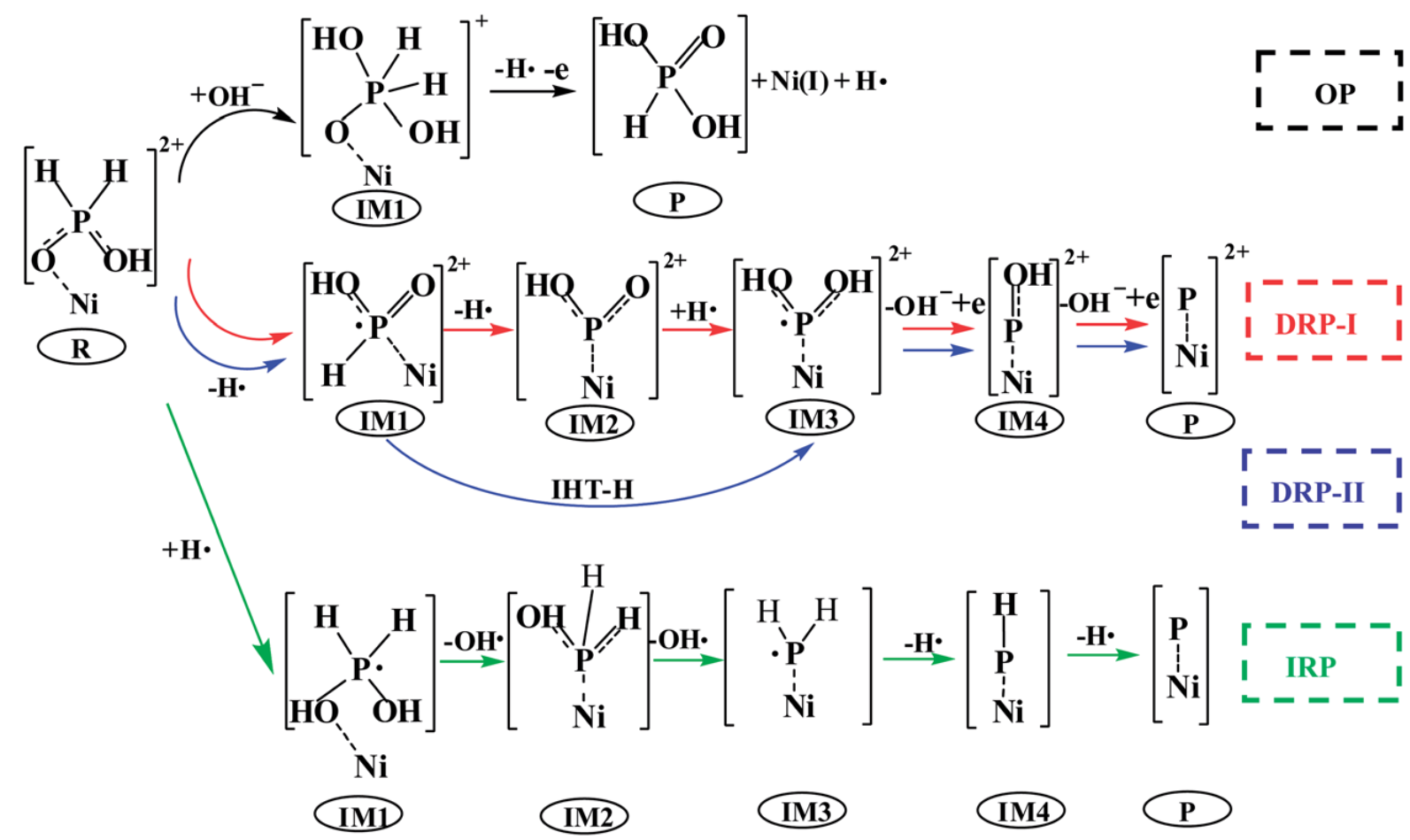
示意图1. 次磷酸的氧化还原耦合途径:包括黑线所示的氧化途径(OP)、红线所示的直接还原途径(DRP-l)和蓝线所示的直接还原途径(DRP-11)、绿线所示的间接还原途径(IRP)。
DFT分析反应路径的可行性
次磷酸氧化路径和通过OP路径的势能分布图分别如示意图1和图3所示。在OP路径中,次磷酸与OH–结合,首先形成IM1。在此过程中,TS1的相对能量为186.5 KJ mol-1,是整个路线中势垒最高的能量。随后,IM1化合物失去一个氢自由基和一个电子。在从IM1到产物的反应中,能量势垒是91.5 KJ mol-1。显然,最高的能量势垒(236.9 KJ mol-1)是TS1的形成过程带来的,其计算值比Homma模型低。能量势垒的降低可以归因于Ni(II)和Ni9团簇的催化作用。
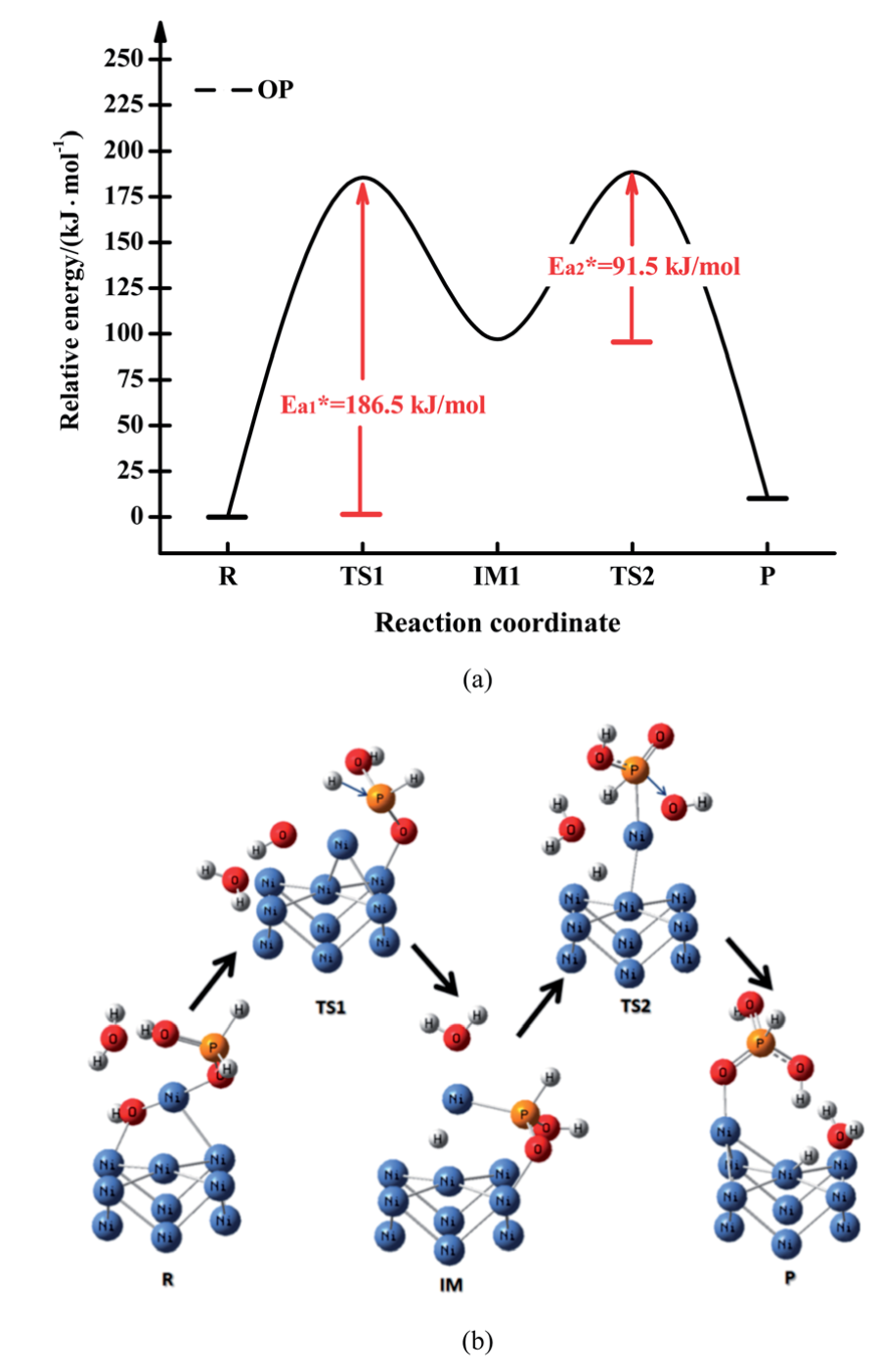
图3. 通过OP路径氧化次磷酸的势能分布图(a)和优化的几何形状(b)。在B3LYP/6-311G(d.p)水平上获得的能量数据。(颜色:Ni-蓝,O-红,P-橙,H-白;蓝色箭头-振动方向。IM =中间态,TS =过渡态。所有的能量都与起始物质有关。蓝色箭头表示过渡态的虚频率处的振型。)
在TS1的形成过程中,Ni(II)较好地吸引了[Ni-(H3PO2)]2+中的氧原子上的电子云。这使得[Ni-(H3PO2)]2+中磷原子的正电性增加。此外,磷原子较高的电性有利于与OH–的结合。IM1的详细电荷分布如图7a所示。
通过DRP-I直接还原次磷酸的路径如示意图1和图4所示。首先,P-H键断裂,同时Ni(II)与磷原子成键。在IM1形成过程中,TS1的能量势垒为42.9 KJ mol-1。然后,另一个P-H键断裂,P=O键上的氧原子被氢自由基氢化。随后,P-(OH)2结构形成了IM3。在DRP-I反应路径中计算出的最高能量势垒未132.3 KJ mol-1,与IM3的形成相关。最后,P-(OH)2通过两步脱水反应被氢攻击形成了P和两个H2O。

图4. 通过DRP-I途径还原次磷酸的势能分布图(a)和优化几何形状(b)。在B3LYP/6-311G(d,p)水平上获得的能量数据。(颜色: Ni-蓝,O-红,P-橙,H-白; 蓝色箭头-振动方向。IM=中间态,TS =过渡态。所有的能量都与起始物质有关。蓝色箭头表示处于过渡状态时的主频率处的振动模式。)
通过DRP-II路径直接还原次磷酸的路径如示意图1和图5所示。与DRP-I不同,P=O键通过从磷原子到氧原子分子间转移氢转变为P-OH。具体来说就是,P-H键首先断裂,然后氢从磷原子转移到氧原子,导致P=O键的断裂。在此过程中,计算得到的能量势垒为284.6 KJ mol-1。然后,P-OH (IM4)键断裂,形成一个被吸附的磷原子。
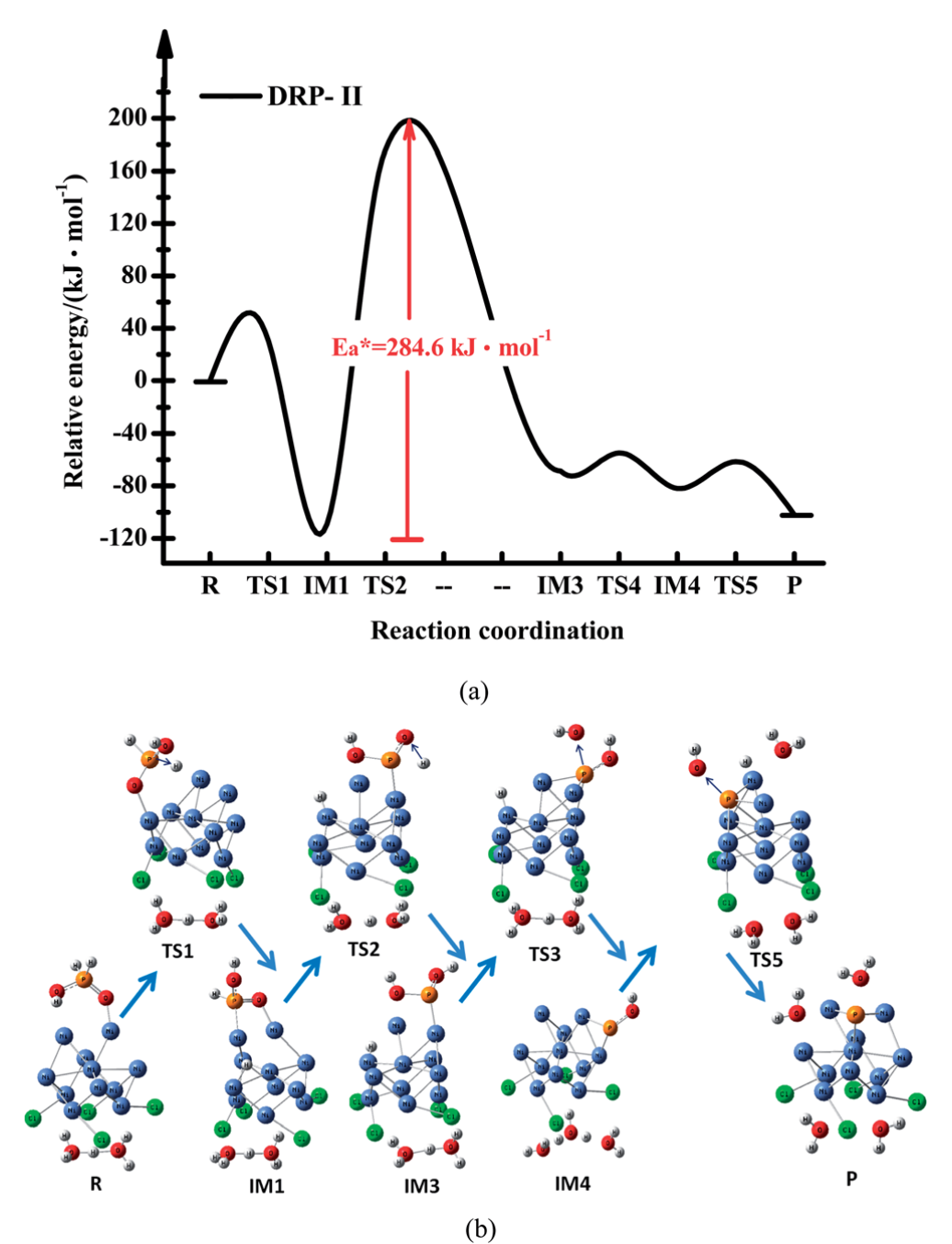
图5. 通过DRP-Il途径还原次磷酸的势能分布图(a)和优化几何形状(b)。在B3LYP/6-311G(d,p)水平上获得的能量数据。(颜色: Ni-蓝,O-红,P-橙,H-白;蓝色箭头-振动方向。IM =中间态,TS =过渡态。所有的能量都与起始物质有关。蓝色的箭头表示在过渡中的主频率处的振动模式
通过IRP路径的间接还原反应路径如图1和图6所示。首先,当氢自由基与氧原子结合时,P=O键断裂。在此过程中,能量势垒约为225.9 KJ mol-1。这两个双P-OH键相继断裂,释放出两个羟基自由基。最终,P-H (IM3)断裂并释放两个氢自由基。原则上,在克服TS1后,次磷酸可以迅速还原为磷原子。
显然,DRP-I通路的能量屏障远低于DRP-Il和IRP。因此,DRP-I反应路径被认为是次磷酸氧化还原的主要还原路径。同时,IRP的能量势垒接近OP (186.5 vs. 225.9 KJ mol-1)。因此,在电化学过电位测量中很难区分这两个反应。
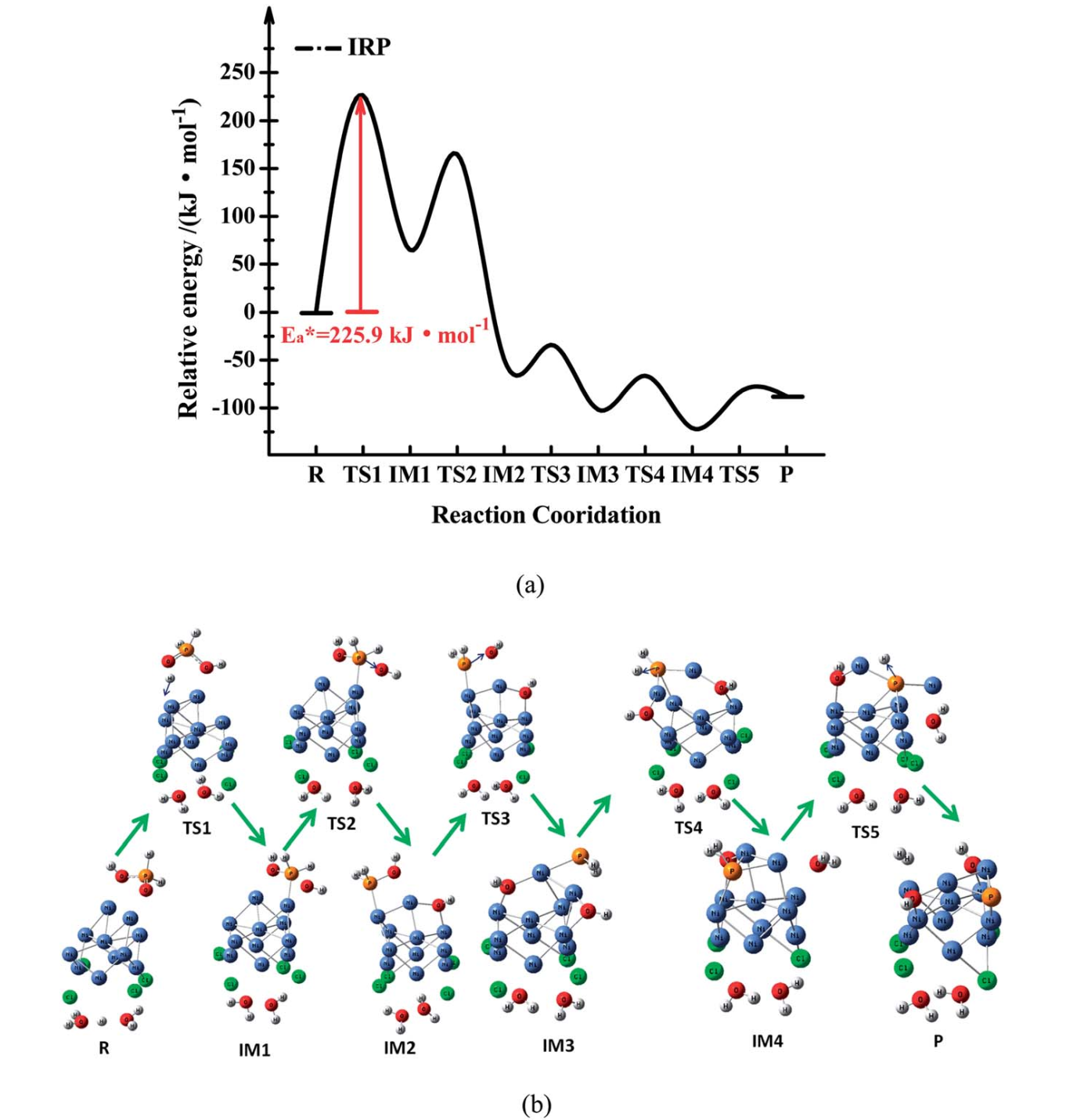
图6. 通过IRP途径还原次磷酸的势能分布图(a)和优化几何形状(b)。在B3LYP/6-311G(d,p)水平上获得的能量数据。(颜色: Ni-蓝,O-红,P-橙,H-白; 蓝色箭头-振动方向。IM =中间态,TS =过渡态。所有的能量都与起始物质有关。蓝色的箭头表示在振动状态下的虚频率处的振动模式。)
图7显示了氧化还原路径的最高位垒处的临界过渡态。在OP-TS1中,氧化过程中最高的位垒是OH–与次磷酸的结合。磷原子和氧原子的计算电荷分别为1.737和-1.086。它们的电荷差是2.818,这个过程的势垒是186.5 KJ mol-1。
(DRP-I)-TS3、(DRP-II)-TS2和(IRP)-TS1是还原路径的最高位垒。在这三种过渡态中,氢原子与氧原子结合,打断了P=0键。图7b-d中氧与氢的电荷差分别为1.312、1.009、1079。另外,对于(DRP-I)-TS3、(DRP-II)-TS2和(IRP)-TS1,相应的能垒分别为132.3,284.6和225.9 KJ mol-1。因此,在P=0过程中,较大的电荷差有利于减小能量势垒。

图7. 包括Ni9簇和Ni(II)离子在内的OP、DRP-1、DRP-ll和IRP通路的最高位垒区TS1、TS3、TS2和TS1的NBO电荷分布。主族元素原子在B3LYP/6-311G(d.p)水平,而镍在LANL2DZ水平。(颜色: Ni-蓝,O-红,P-橙,H-白;橙色箭头-振动方向。)
次磷酸分解的EIS分析
图8显示了不同pH值下化学镀镍溶液的奈奎斯特阻抗谱。pH值以步长2.0从3.0增加到9.0。在pH=3.0溶液中,有两个典型的时间常数,分别对应于高频段和低频段的两个独立的电容阻抗回路。在pH=3.0时,样品不能通过化学镀镍沉积。当pH值增加超过5.0时,在中频域中出现一个感应阻抗弧,弧的半径随着溶液中pH值的增加而减小(图8b)。当pH值从5.0增加到9.0时,第一个电容阻抗回路在高频区域从4.42 cm逐渐增加到8.60 cm2。
图9为R (Cdl1Rt1)(ZRt2)(Cdl2Rt3)的等效电路模型,用于模拟EIS谱。在等效电路模型中,Rs表示电解液的电阻。在高频域中存在一个典型的电容性弧(CL-H),由Cdl1和Rt1并联模拟所得。在中频区域有一个电感弧(IL-M),由Z和Rt2并联组成。在低频区域也有一个电容弧(CL-L)区域,表示Cdl2和Rt3并联。图9b为实验数据与模拟结果的对比图,拟合良好。Nyquist图是在沉积电位第一个小时的pH=5.8下得到的。图10为不同pH值和沉积时间下化学镀镍的Nyquist阻抗谱。在pH=5.0时,在奈奎斯特图中观察到三个时间常数,包括两个电容弧和一个电感弧。当pH值从5.0增加到9.0时,CL-H环在高频区域的直径从5.3增加到25.4 Ω cm2。这些结果与Touhami的研究结果一致,表明电极表面吸附了羟基。然而,结合DFT结果,(CL-L)弧会与Ni-P化合物发生反应,后者会紧密地吸附在Ni表面。同时,它们对羟基很敏感,而羟基可以与氢自由基结合,导致化合物浓度下降。该结果表明,磷的掺杂比例随pH值的增加而降低。同时,电感(IL-M)弧变得更明显。此外,CL-L弧的直径在低频区域从6.5增加到13.2 Ω cm2。值得注意的是,由于反应物浓度(如Ni(II)和H2PO2-)的降低以及产物浓度(如H3PO3-和H+)的增加,三种电化学过程的电阻随着沉积时间的增加而逐渐增大(表1)。

图8. pH值分别为3.0、5.0、7.0和9.0 (a)和部分放大图 (b)溶液中EN沉积的Nyquist阻抗谱。
表1. OP、DRP-I、DRP-Il和IRP的最高能量势垒
| Pathways | OP | DRP-I | DRP-II | IRP |
| The highest energy barrier/KJ mol-1 | 186.5 | 132.3 | 284.6 | 225.9 |
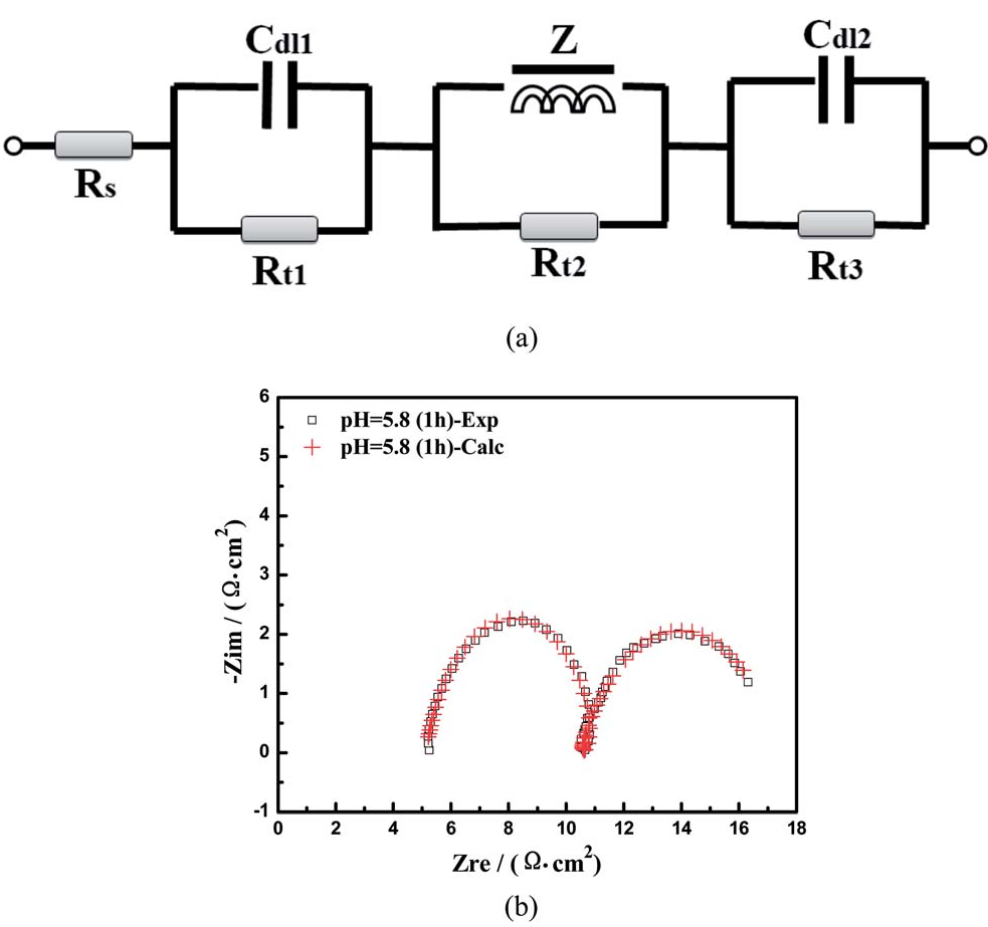
图9. 等效电路模型(a),以及在EN过程的第一个小时内从pH=5.8槽中模拟和测量的阻抗谱(b)。
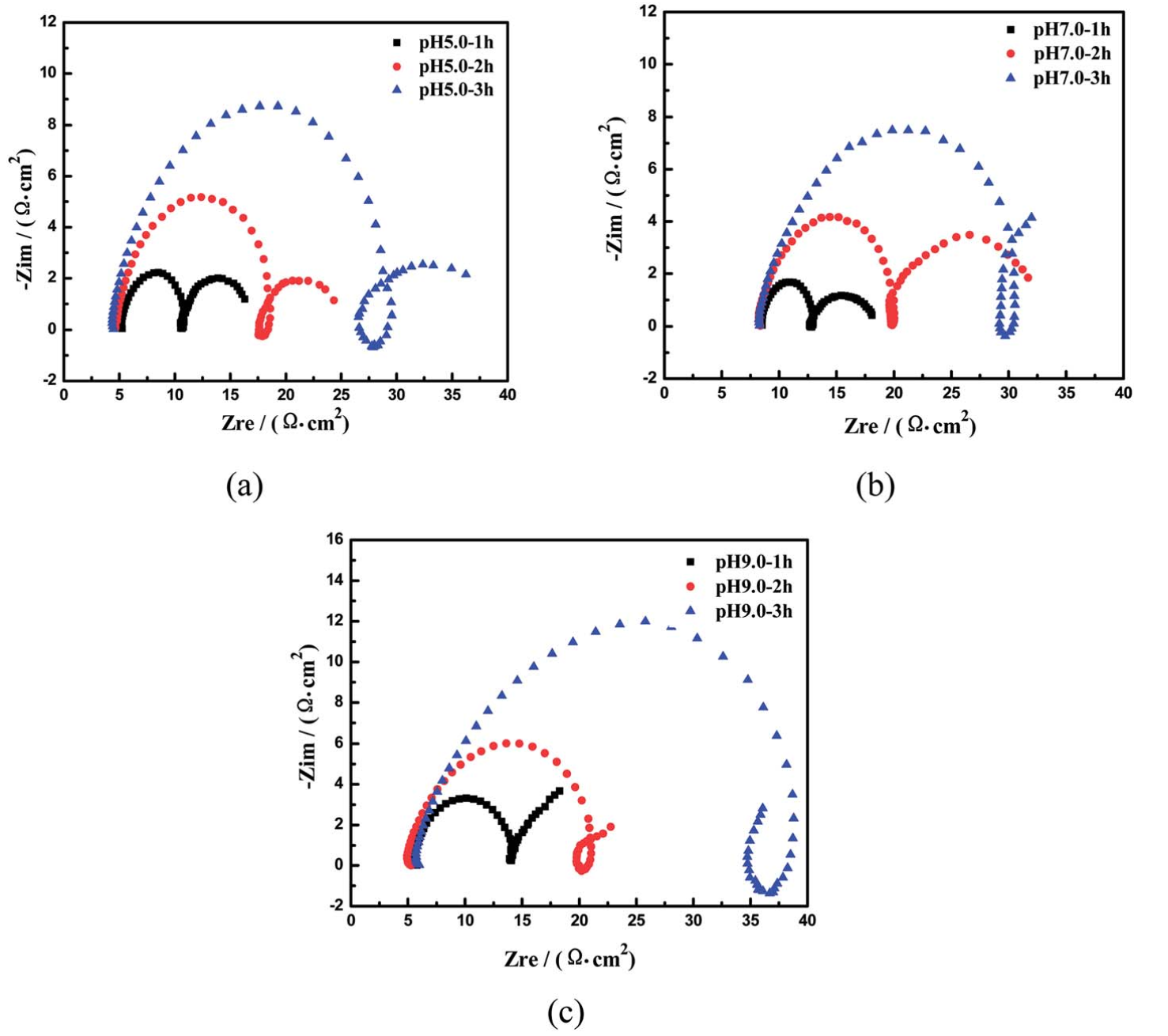
图10. 不同反应时间下,pH值分别为5.0 (a)、7.0 (b)和9.0 (c)溶液中化学镀镍的Nyquist图。
次磷酸的催化机理
结合DFT和EIS的结果,化学镀镍最有可能通过OP和DRP-I路径进行。它们包括三种典型的电化学过程,即双电层的充放电过程、不稳定中间体对镍的吸附过程和Ni-P化合物中间体对镍的吸附过程。其电化学过程和相关的种类列举在Table 2。
表2. Nyquist图与界面反应特征的关系
| Frequency domain | Electrical Element | Interfacial reaction characteristic | Related Species |
| High | 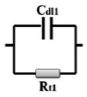 |
Charge–discharge in electric double layer | Ni2+, H2PO2–, H3PO3–, H+, OH– |
| Middle | 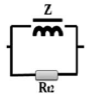 |
Adsorption of unstable intermediate on nickel surface | Ni(I) |
| Low | 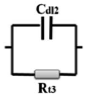 |
Adsorption of Ni–P compound intermediates on nickel surface | OP-IM1, H RP-IM2, IM3, IM4, P |
基于DFT分析,次磷酸的偶联氧化还原反应机理如示意图2所示。在化学镀镍过程中,Ni(II)获得两个电子,并逐渐向Ni(I)和Ni(0)转变。当其与OH–结合时,电子来自氢自由基。在次磷酸的氧化过程中,[H3PO2]与一个OH–结合,释放出一个氢自由基。因此,较高的OH–浓度有利于氧化反应的进行。另一方面,在次磷酸还原过程中,随着[H3PO2]逐步释放两个氢自由基,[HPO2]2+能够捕获一个氢自由基,形成[H2PO2]2+。然后[H2PO2]2+释放两个羟基自由基,导致磷掺杂到镍镀层中。很重要的是,当次磷酸转化为次磷酸离子时,还原过程需要一个氢自由基。氢自由基在相对较低的pH值环境中更稳定,因为它们倾向于与OH–结合。因此,还原过程将在较低的OH–浓度下进行。总的来说,OH–是控制次磷酸氧化还原反应的关键物质。在较低pH的溶液中容易获得高P掺杂的镀层。
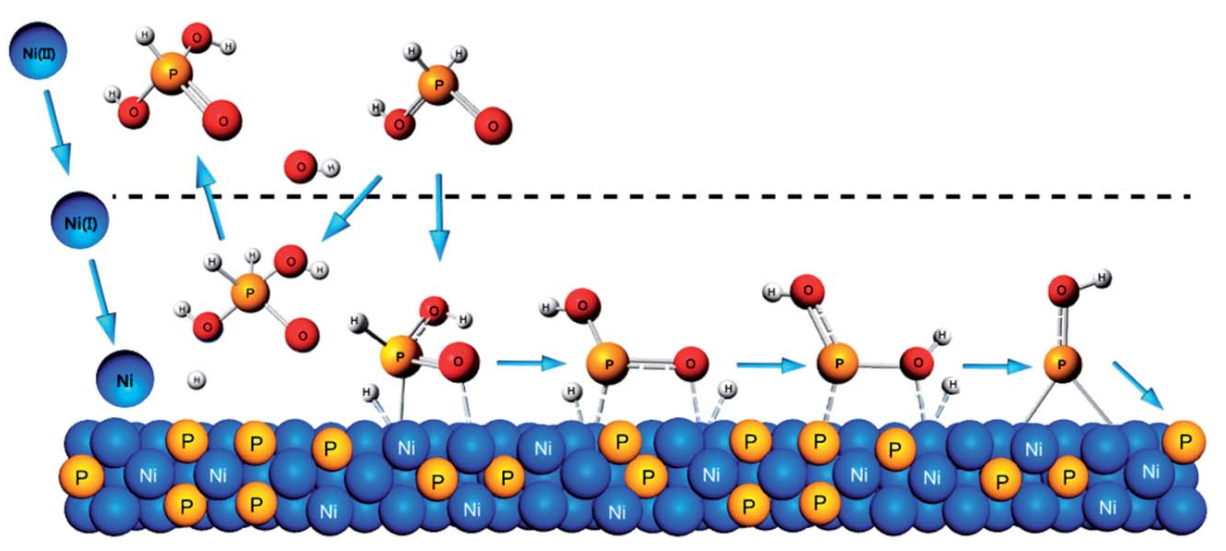
示意图2. 次磷酸化学镀镍在镍表面的耦合氧化还原反应机理。
结论
本工作为获得高磷非磁性化学镀镍涂层提供了一种思路,该涂层可作为硬盘驱动的表面涂层。在化学镀镍过程中,磷的掺杂量随着pH值的降低而增加。当pH值低于5.0时,磷含量超过13.8 wt%,镀层开始变得无磁性。为了阐明磷的掺杂机理,采用DFT的方法分析了次磷酸的耦合氧化还原反应路径。确定了最可能的氧化还原反应路径为OP和DRP-I。此外,进一步借助ElS的方法对电化学镀镍的工艺进行阐述。特别地,根据所获得的EIS结果,反映出三个电化学反应过程。电化学阻抗谱和DFT相结合的研究为研究磷含量可控的化学镀镍机理提供了新的思路。此外,还讨论了OH–在非均相电催化过程中抑制磷掺杂的机理。
理论计算
所有的DFT计算都是使用Gaussian 03进行的,其中使用了混合的Becke 交换,Lee, Yang和Parr correlation (B3LYP)密度泛函方法。对于氢、氧、氯和磷,本工作使用6-311G(d,p)基组。对镍原子的有效核电位(ECP)的计算采用LANL2DZ基组。这些基组对研究无电化学和其他电化学过程的机理是有效的。通过计算各组分的分子基态几何和振动频谱,确定了反应路径势能面。静态计算中不存在虚频,而在过渡态中只确定一个虚频。虚振动模态的可视化检查采用Gaussian View 3.7程序包。此外,采用显式溶剂化模型研究了水溶液的溶剂化效应。在该模型中,Ni(II)离子与两个水分子形成络合物。与Homma之前的模型不同,我们的反应模型是在镍团簇表面同时存在次磷酸盐离子和Ni(II)离子的封闭系统。


