
摘要:类先导药物或分子量300以下的药物是当前药典和现代药物化学实践中一个重要的、有时被忽视的组成部分。为了研究类先导药物发现的最新进展,我们分析了在2011年至2017年间批准的药物和前200名的处方药物,并提供了最近批准的类先导药物的案例研究。在这些案例中,有许多药物与之前已知的药物或天然底物类似,其药物化学优化的关键点之一是选择低分子量的起点,并在优化过程中保持低分子量。然而,低分子量起点的识别受到合适的低分子量筛选集可用性的限制。为了提高类先导药物的发现速度,我们建议在筛选库中增加对类先导起点的关注。
编译:李凯丽,高银谊,柏川/中山大学医学院
日期:2020-03-16
原文:Raymer, B.; Bhattacharya, S. K. Lead-like Drugs: A Perspective. J. Med. Chem. 2018, 61 (23), 10375–10384. https://doi.org/10.1021/acs.jmedchem.8b00407.
在瞬间我们可以领略纯美, 在刹那生命可以精彩。
–Ben Jonson. Underwood (1640)
介绍
类先导药物(Lead-like drugs)或分子量小于300的药物是当代药典和药物化学重要的但有时却被忽略掉的组成部分[1,2]。2011年,Walters及其同事整理了50年内批准药物的理化性质。他们发现早期(1959-1964年)批准药物的分子量范围主要为300-360,后随着时间的推移,分子量逐渐增加,至2005-2009年获批药物分子量主要为360-440。综上得出的合理结论是,类先导药物的平均分子量通常在300−440之间。类先导药物的分子量以300作为界限,这也符合三法则(rule-of-three,Ro3)的分子量标准,尽管这个标准主要是用于基于片段的药物的片段选择上[3]。考虑到这些范围和分子量变化的趋势,我们建议将分子量﹤300的化合物认为是非典型药物,并使用这个标准作为类先导药物的上限[4]。
为了研究类先导药物发现的最新进展,我们调查了2011年至2017年间[美国食品和药物管理局(FDA)和欧洲药物管理局(EMA)]的药物批准情况(图1;完整的药物清单请查阅补充信息),但不包括分子量大于2000的药物。我们还分析了2014年美国处方数据中排名前200的处方药(品牌药和非专利药)[5]。这个数字突出了最近批准的类先导药物的数量,此外,还强调了基于前200名处方药处方水平的类先导药物的长期成功。在2011年至2016年期间,近期批准的146种药物中有25种(17%)的分子量是低于典型分子量范围300-440。在排名前200位的处方药中,160种低分子量药物中有57种的分子量低于300,占处方低分子量药物的36%。值得注意的是,从2011到2016年间批准的药物中占比例较大的分子量大于400的药物,与在前200名处方药物中所占比例较小分子量大于400的药物有所不同。在比较2011-2016年批准的药物和前200种处方药物时,批准的药物中分子量低于300、分子量在300-440之间和分子量大于440的相对比例有显著差异。在前200名处方药物中,分子量在300以下的药物占很大比例;而在2011 年至2016年间,批准的药物中分子量在440以上的药物所占比例更高。许多排名前200位的处方低分子量药物已经上市很长时间了;鉴于最近批准的分子量分布,观察高处方药物的分子量分布是如何随时间演变将是一件有趣的事情。在2017年,因为FDA批准的28种药物中有18种的分子量大于440,而只有3种的分子量小于300,故高分子量药物所占比例更大的趋势仍在持续。
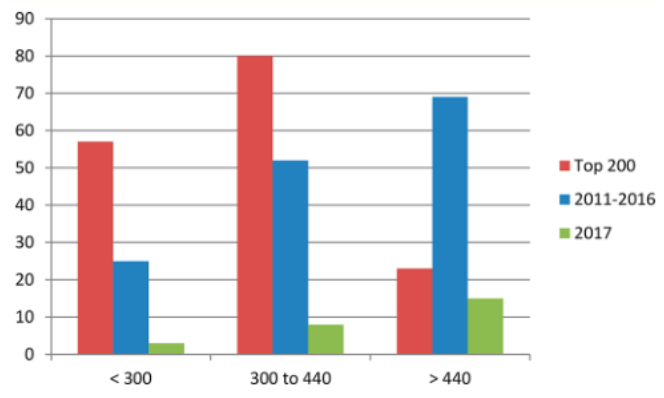
Figure 1. 新批准的小分子药物与处方量大的药物的数据,直方图按分子量范围取间隔。红色:2014年美国处方量最高的200种药物;蓝色:2011-2016年美国批准的药物;绿色:2017年美国批准的药物。
虽然药物化学家们对化合物分子量的变化趋势很感兴趣,但发现这些药物背后的故事和通用原理的应用在开展当前和未来的项目中都很有价值。在本文,我们精选了一些最新获批的新药进行案例分析并对共同的主题(表1)进行讨论[6]。我们将类先导药物分为三类:由类先导化合物筛选产生的;通过药物化学优化而来的;通过生物学和临床试验(也就是说药物老药新用、代谢或者天然产物)获得的。通过对这些案例进行分析,我们讨论了类先导药物的ADME和毒性特征,并基于这些常见的话题对类先导药物的开发提出了建议。
表1. 用做案例研究的药物精选自2011-2017年间批准的药物a
案例研究:来源于类先导化合物筛选工作的药物
Tavaborole (1, Kerydin)
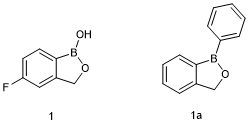
他伐硼罗(Tavaborole,商品名Kerydin)通过抑制真菌细胞质亮氨酰转移核糖核酸(tRNA)合成酶发挥局部治疗指(趾)甲甲真菌病(灰指甲)的作用[7]。它的发现首先是对含硼小分子化合物库进行抗菌试验的筛选,确定了含硼化合物(1a)[8]。后对筛选出来的化合物进行药物化学方面的研究,主要是在分子结构的苯环上加入氟取代基和用苯基取代硼基团上的羟基,确定了他伐硼罗(1)的结构[9]。综合指甲渗透和角蛋白结合临床前研究的结果,他伐硼罗被确定为临床候选药物展开进一步的评估。他伐硼罗的研究案例突出了类先导化合物筛选工作的实用性,一个是利用合适的体外测定手段对类先导小分子化合物库进行筛选能够实现高效的化合物开发,一个是可以采用局部方法来避免系统安全的问题。
Crisaborole (2, Eucrisa)
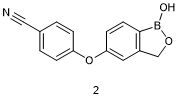
含硼化合物能够抑制细胞因子,如肿瘤坏死因子(TNFα)和α干扰素(IFNα)从外周血单核细胞的释放,并且这类化合物被发现还是PDE4酶抑制剂[10]。类似于上述他伐硼罗的发现,对含硼小分子化合物库进行筛选并结合随后的药物化学改造,开发了候选化合物,AN-2728,后命名为克立硼罗(crisaborole,商品名Eucrisa)。克立硼罗是首个获得美国FDA批准的PDE4酶抑制剂,适用于两岁及以上年龄过敏性皮肤炎患者。克立硼罗对多种PDE4亚型(PDE4A1A, PDE4B1, PDE4B2, PDE4C1, PDE4D7)具有较强的抑制活性(IC50 = 55-340 nM),对其他PDE同工酶的抑制活性较弱。克立硼罗分子中硼原子的四面体结构与环磷酸腺苷(cAMP)中的磷酸盐相似,被认为与PDE4酶中cAMP的结合位点发生了重叠。
Ataluren (3, Translarna)
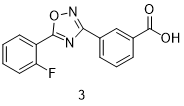
氨基糖苷类抗生素可修饰基因表达,并且能够抑制破坏核糖体正常功能的终止密码子。然而,它们往往是非特异性的,从而导致严重的副作用。据报道,小分子阿塔卢伦(Ataluren,3,商品名Translarna)能以较低的毒性来调节细菌核糖体。两种针对低分子量化合物的高通量筛选(HTS)确定了促进终止密码子UGA无义抑制(nonsense suppresion)的调节剂。恶二唑系列从先导优化鉴定的多类化合物中筛选出来的。恶二唑和苯甲酸部分对活性至关重要,而且羧酸部分与mRNA中的一个氨基的H键连接已被证明是一个重要的相互作用[11-13]。尽管阿塔卢伦已在欧盟获得有条件的批准,但在美国,它尚未被FDA批准,并且被要求进行更多的临床试验。
Vortioxetine (4, Trintellix)
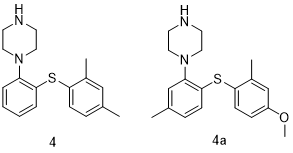
沃替西汀(Vortioxetine,4,商品名Trintellix)是一种5-羟色胺调节剂,被批准用于治疗重度抑郁症(MDD)。发现沃替西汀的研究的基本目标是发现可同时作用于血清素转运体(SERT)和5-HT1A受体上的新一代抗抑郁药物[14]。一个有针对性的筛选项目确定了一个芳基哌嗪化合物(4a),而4a最终转化为4(沃替西汀)。沃替西汀具有多种药理作用,具体表现为5-HT3A和5-HT7受体的拮抗剂、5-HT1B受体的部分激动剂、5-HT1A受体的激动剂,以及SERT抑制剂。这种协同的多模式5-羟色胺能活动可能导致更强的抗抑郁作用。
案例研究:来源于已知的类先导底物与药物
Edaravone (5, Radicava)
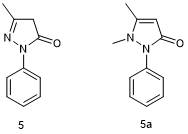
2017年美国FDA批准依达拉奉(Edaravone,商品名Radicava,5)以静脉输注的给药方式用于治疗肌萎缩侧索硬化(Amyotrophic Lateral Sclerosis,ALS),这是20多年来首个FDA批准用于治疗ALS的药物。虽然依达拉奉治疗ALS的作用机制尚不明确,但自20世纪80年代以来,依达拉奉就已经开始作为一种脑保护剂(自由基清除剂)在使用,并且先前在日本被批准用于治疗急性脑梗死[15]。依达拉奉(MCI-186)在体外可抑制非酶脂质过氧化和降低脂氧合酶活性,在体内可减轻缺血性脑肿胀和缺血后脑肿胀[16]。依达拉奉由安替比林(Phenazone,5a)衍生而来,1887年首次合成的安替比林,是一种镇痛药和非甾体抗炎药(NSAID)[17]。起初的研究表明,依达拉奉对有类似ALS症状的小鼠有效,后来经临床试验确定该药可用于治疗ALS并且依达拉奉的口服制剂已经进入临床开发阶段。
Tasimelteon (6, Hetlioz)
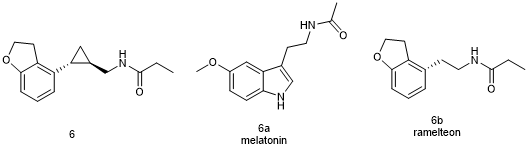
他司美琼(Tasimelteon,商品名Hetlioz,6)对褪黑素MT1和MT2受体都具有亚纳摩尔级别的效价,是一种选择性的褪黑素受体激动剂[18]。该药主要用于治疗盲人的非24小时睡眠觉醒障碍(non-24h sleep-wake disorder)。他司美琼是天然底物褪黑素(Melatonin,6a)的结构类似物,同时也是另一种褪黑素激动剂雷美替胺(Ramelteon,6b)的类似物,雷美替胺也具有选择性地与MT1和MT2受体结合的作用。与天然底物褪黑素相比较,他司美琼不仅具有更优的药代动力学性质,还能避免色氨酸的犬尿代谢物的形成[19]。在这个案例中,我们可以看到,作为类先导药物,他司美琼的发现得益于利用药物化学的手段对现有的结构类似物进行优化,在这里与他司美琼结构类似的药物与中枢神经系统胺能G蛋白偶联受体(GPCRS)靶点的天然底物有关。
Lorcaserin (7, Belviq)
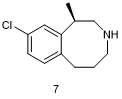
众所周知,激活中枢5-羟色胺(5-HT)能通道可以降低食欲和减轻体重[20]。值得注意的是,激活非选择性5-HT2C受体能够显著地减轻体重,但同时也存在着严重的毒性问题,比如心脏瓣膜缺陷增加。为了减轻毒性脱靶效应,药物化学家们重点研究将5-羟色胺能化合物骨架的构象加以限制,以期能提高药物对5-HT2C受体的选择性,降低药物对5-HT2A和5-HT2B受体的作用[21]。在起初的实验室试验中,氯卡色林(Lorcaserin,商品名Belviq,7)就显示出体外选择性,良好的体内药代动力学和药效学性质以及显著的疗效成果,随后该药进入了临床试验,在试验中氯卡色林能使肥胖患者的体重持续下降[22]。通过优化现有药物结构得到氯卡色林这一实例,突显了药物化学中构象限制的设计在发现类先导药物上的实用性,并且这一设计可以用来提高药物作用的选择性。
Brivaracetam (8, Briviact)
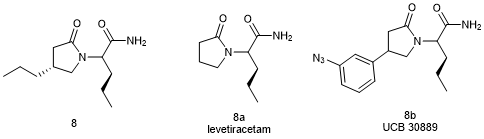
布瓦西坦(Brivaracetam,商品名Briviact,8)的发现源于对抗惊厥和抗癫痫药左乙拉西坦(Levetiracetam,8a)靶点的化学生物学研究[23]。首先利用光亲和探针UCB 30889 (8b)鉴定出突触囊泡糖蛋白2a (SV2A)是药物靶点。随后在药物化学优化中,确定了左乙拉西坦的C2 -丙基衍生物即布瓦西坦作为候选药物[24,25]。与左乙拉西坦相比,布瓦西坦具有更强的药效和更快的起效时间,从而降低了临床有效剂量。发现布瓦西坦的案例显示出了化学生物学和药物化学对优化已有药物的重要性。
Tipiracil (9, as Part of Lonsurf)
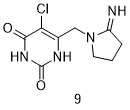
替吡嘧啶(Tipiracil,9)是一种胸腺嘧啶磷酸化酶(TPase)抑制剂,可以阻断该酶的代谢来增加抗癌药物三氟吡啶的相对浓度[26]。替吡嘧啶的发现是从已知的尿嘧啶磷酸化酶(UPase)抑制剂5-卤代-6-氨基尿嘧啶的确定开始的,这是一种与TPase相关的酶[27]。随后,从尿嘧啶衍生物的药物化学优化中发现了替吡嘧啶,该药表现出较好的体外酶抑制作用以及体内药代动力学和药效学,因此可进一步用于人类研究[28,29]。随后的临床试验也证实了替吡嘧啶在提高三氟吡啶浓度方面的功效:与单独使用三氟吡啶相比,合用替吡嘧啶可增强三氟吡啶的功效[30]。替吡嘧啶的发现提示我们可以选择性优化活性药物来作为候选药物。
Pomalidomide (10, Pomalyst)
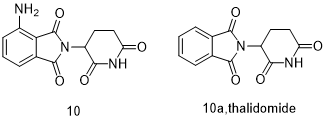
泊马度胺(Pomalidomide,10,商品名Pomalyst)是沙利度胺的3-氨基类似物(10a),适用于多发性骨髓瘤患者[31]。沙利度胺是谷氨酸的合成衍生物,这是一种有争议的镇静药物:由于其致畸性和其他副作用,该药物于20世纪60年代退出市场。沙利度胺后来被证明具有抗血管生成作用,并最终在1998年被批准用于治疗多发性骨髓瘤[32]。来自Dana-Farber的科学家们研究了沙利度胺的3-氨基沙利度胺类似物,发现它比沙利度胺具有更高的TNF-α抑制活性[33],尽管其抗血管生成作用与TNF-α抑制的程度无关。而这两种药物对不同的细胞因子也有不同的调节作用,这些细胞因子可能与骨髓瘤细胞增殖有关。沙利度胺和泊马度胺作为外消旋物出售,这两者在血浆和体内都可以迅速相互转化。泊马利度胺的两种对映异构体均显示出相似TNF-α合成抑制活性。
Pitolisant (11, Wakix)

大脑中的组胺能神经元在维持清醒状态和增强大脑功能方面起着关键作用。替洛利生(Pitolisant,11,商品名Wakix)等药物可以阻断组胺H3受体,进而增强组胺能神经元的活性,提高脑内组胺水平。替洛利生起源于天然底物组胺本身的结构(11a)。Thioperamide (11b)和ciproxifan (11c)是以前发现的另外两种强效H3受体拮抗剂,但由于具有肝毒性而不能进行下一步研究。最终选用哌啶环代替咪唑环得到了替洛利生[34]。
案例研究:来源于生物学和临床试验研究的药物(老药新用、药物代谢或者天然产物)
Dimethyl Fumarate (12, Tecfifidera)
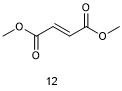
第一次尝试使用富马酸来治疗炎症的是一位德国生物化学家,他自行服用富马酸,通过纠正自身的富马酸不足来治疗他的牛皮癣[35]。富马酸二甲酯(Dimethyl Fumarate , 商品名Tecfifidera,12)是富马酸的二酯前体,富马酸是柠檬酸循环的代谢组分。从这个角度看,富马酸二甲酯是一种内源性人体代谢物的前药。最初,富马酸二甲酯用于细胞的辐射增敏[36]。随后对富马酸酯组合物的研究表明,富马酸二甲酯可用于治疗牛皮癣和多发性硬化症。在一项临床3期试验中,富马酸二甲酯作为单一药物使用能够显著降低多发性硬化疾病的活跃程度[37,38]。最近的研究表明,富马酸二甲酯通过抑制泛素链的形成,从而发挥阻断细胞因子产生的作用[39]。鉴于富马酸二甲酯的结构人们都很熟悉,富马酸二甲酯是基于它的制剂和使用方法的专利来商业化。值得注意的是,富马酸二甲酯也可以作为家具治疗剂来防止霉菌生长,但由于存在接触性皮炎的风险,这一应用已经被停止[40]。富马酸二甲酯治疗多发性硬化症的进展突显了偶然性发现药物和药物老药新用的可能性。
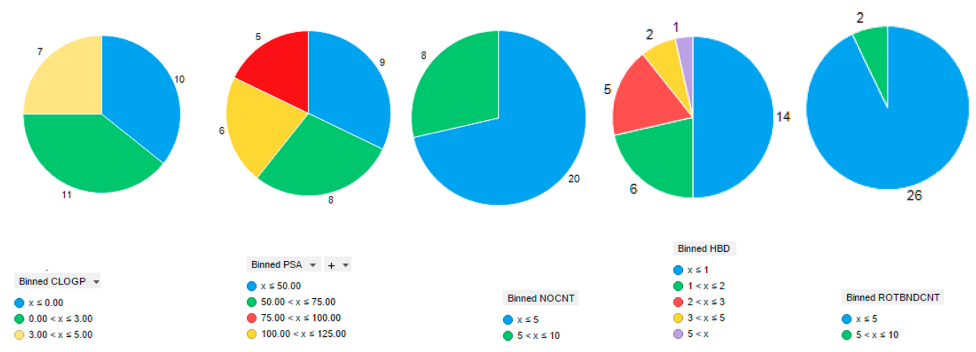
Figure 2.2011-2017年间批准上市分子量小于300药物的油水分配系数(clogP)、极性表面积(polar surface area,PSA)、氮与氧原子数(NO)、氢键供体数(hydrogen bond donor,HBD)、柔性键数(rotational bonds)
Migalastat (13, Galafold)
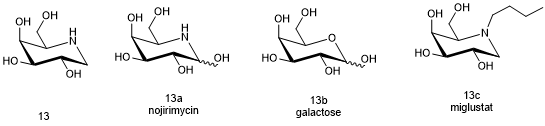
氨基糖类似物米加司他(Migalastat,商品名Galafold,13)(半乳糖抑制素)是从发酵产物中分离得到的,它的结构与诺吉霉素(Nojirimycin,13a)和半乳糖(Galactose,13b)相似[41]。由于米格司他(Miglustat,13c)与α-半乳糖苷酶A(α-galactoside A)的底物的末端半乳糖-酰基鞘胺醇三己糖(Globotriaosylceramide)结构相似,人们认为米格司他这个天然产物很有可能成为α-半乳糖苷酶A的调节剂[42]。体外和体内实验表明,米加司他可以作为α-半乳糖苷酶A的药物伴侣,保护酶的活性,减少酶底物的形成[43,44];而且在此之前,类似的底物已经被用作酶药物伴侣在使用[45]。在酶替代治疗法中,β-半乳糖苷酶A冻干粉注射剂(Fabrazyme)和基因重组半乳糖苷酶阿尔法注射剂(Replagal)两种酶药物用于治疗法布里病(Fabry disease)[46,47]。而在药物伴侣法中,利用口服药物模拟上述两种酶药物的疗效。米加司他每隔一天给药150毫克,可以增加患者α-半乳糖苷酶A的活性,减少酰基鞘胺醇三己糖的积累[48]。综上所述,使用类先导天然产物来模拟疾病中的反应底物已经被证明是治疗法布里病的有效方法。
Pirfenidone (14, Esbriet)
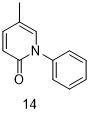
抗纤维化化合物吡非尼酮(Pirfenidone,14,商品名Esbriet)可以减少成纤维细胞增殖和胶原蛋白的产生,因此被用于治疗特发性肺纤维化(IPF)[49]。在20世纪70年代吡非尼酮首先被当做抗炎药进行研究,在临床前研究显示该化合物对肺成纤维细胞有积极作用后作为IPF治疗用药[50]。临床试验表明,吡非尼酮在IPF治疗中具有良好的收益-风险关系;而对吡非尼酮临床试验的meta分析表明,在临床试验中吡非尼酮与降低死亡率的相对风险有关[51,52]。吡非尼酮作为抗纤维化药物可能有多种用途,比如单独使用或与I型血管紧张素受体拮抗剂联合使用[53,54]。吡非尼酮深入利用显示出可信的临床前生物学研究可有力推动候选药物的老药新用。
Teriflunomide (15, Aubagio)
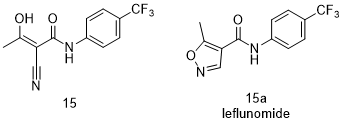
特氟米特(Teriflunomide ,15,商品名Aubagio)是来氟米特(leflunomide,15a)的主要活性代谢物,而来氟米特是一种治疗类风湿性关节炎的药物,分别于1998年和1999年在美国和欧洲获得批准。来氟米特最初是一种农业杀菌剂,后来被发现是一种抗炎免疫调节剂。口服的来氟米特会迅速转化为其主要活性代谢物(15)[55,56]。特氟米特已被证明可以通过结合到二氢硼酸脱氢酶(DHODH)的一个狭窄疏水性通道这一活性位点,进而抑制DHODH。
Eslicarbazepine Acetate (16, Aptiom)
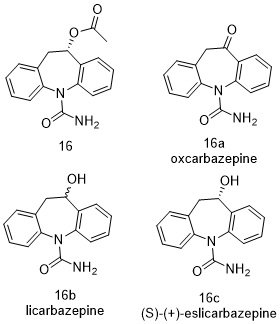
依卡西平(Eslicarbazepine Acetate,16,商品名Aptiom)是一种电压门控钠通道阻滞剂,已被批准用于部分发作性癫痫病。它与20世纪90年代作为低分子量抗癫痫药物引入的奥卡西平(oxcarbazepine,16a)有关[57]。经测定,奥卡西平可在体内迅速而广泛地代谢为相应的还原醇(16b、C10-S和C10-R,比例为4:1)的外消旋混合物。这两种醇都被分别合成,同时还合成了一系列可作为前药的酯类。对这批化合物的研究表明,S -乙酸盐(16,eslicarbazepine acetate)在体内外都具有最高的钠通道抑制活性。尽管人钠通道受体的晶体结构这一问题尚未得到解决,但据推测,亚型非选择性钠通道调节剂如eslicarbazepine(16c)也许与细胞内某种钠通道孔内的位点结合。
讨论
在对2011年至2017年间批准的174种类先导药物中的28种的检查中(请参阅补充信息),发现这些化合物的理化性质都介于“三法则”和“五法则”之间(图2)。例如,在分子量低于300 的28种化合物中,有21种化合物的log P值小于或等于3,20种化合物具有3个或更少的氢键供体,18种化合物具有3个或更少的氢键受体。当我们从“五法则”的角度来看,在这28种化合物中只有1种是具有6个氢键[58]。此外,在2011年到2017年间批准的所有类先导药物的总极性表面积均低于125 Å2,可旋转键数均低于10,这些都符合口服生物利用度的性能范围[59]。
在对最近批准的分子量低于300的化合物的案例研究时,发现有三个特点。首先,在这些分子量低于300的化合物中,有一部分化合物来源于类先导药物的筛选,如他伐硼罗、克立硼罗和阿塔卢伦。其次,最近批准的药物中有很大一部分是基于药物化学优化,或者是已知类先导药物以及天然产物的衍生化。例如,氯卡色林是已知的5-羟色胺能骨架构象受限的类似物,他西美汀与已知的褪黑激素受体激动剂有关,而泊马度胺是沙利度胺的结构类似物。从前面两个特点点中,可以看出药物化学起点的分子量对被批准的化合物分子量有很大的影响[60,61]。第三,在最近批准的类先导药物中,老药新用化合物(如富马酸二甲酯、吡非尼酮)和活性代谢物占了很小但是很重要的一部分。值得注意的是,虽然案例研究中的类先导药物都有一系列的效价(表1),但在15种药物中,就有11种的配体效率值高于典型化合物(0.45),这与化合物的每个原子的高效价有关[62]。
类先导药物物的ADME与毒性
在理化性质上,类先导药物与已知药物没有显著差异。在上述案例研究中,成功的口服生物利用化合物通常具有两种典型特征,如clogP和极性表面积(请参阅表1)[63]。由于分子量低,这些化合物的配体效率(ligand efficiencies, LE)高于正常范围的上限。但是,这些化合物的剂量范围都在以往药物的使用剂量范围之内[64]。为了进一步研究分子量对ADME的影响,我们分析了105种不同分子量化合物的数据[65]。我们发现,分子量小于300的化合物中有76%的人口服生物利用度大于90%;而分子量大于300的化合物中只有53%的人口服生物利用度大于90%;分子量大于440的化合物中只有42%的人口服生物利用度大于90%。类先导药物的这种生物利用度增加的趋势可能与之前提到的溶解度和渗透性与分子量的关系相一致。具体来说,溶解度往往随着分子大小的增大而降低,而低分子量的化合物可能同时具有细胞旁和细胞外的吸收途径[66-69]。
同样地,Johnson和他的同事对一组47018个不同化合物的分析显示了分子量和log D对清除率和通透性影响的预测值[70]。据预测,分子量低于300的化合物可容许更大范围的亲脂性值(log D为-2至5),同时仍保持体外最高的渗透率和清除率。而且,正如许多报道所言,类先导药物更可能入脑分布[71]。对案例研究药物的半衰期值和分布容积的检验表明,这些值与典型的上市药物的值一致[72,73]。分子量也可能影响高分子量药物的排泄途径:分子量增加有利于胆汁排泄,分子量减少有利于肾脏排泄,这可能是由于转运体对清除的影响增加导致的[74-76]。这些案例研究化合物反映了类先导药物可能通过肾脏排泄,因为在尿液中发现了这些化合物的母体或代谢物(表1)。
关于分子量对毒性的总体影响的研究较少。根据先前的分析,分子量低于300的化合物可能可以降低毒性风险[77]。通过对50000个小分子化合物的编辑和分析,Struck、Schmidt和他们的同事发现可以将毒性和分子量与毒性的实验测量值联系起来[78]。具体而言,与分子量在300-400或在400-500相比,在分子量≦300的化合物中,中等毒性化合物所占的百分比显著降低。更进一步的研究表明,大多数已批准的药物治疗指数(TI)> 10,且游离药物浓度(C eff)> 1μM的化合物,都属于分子量﹤300以及更高极性(clogP﹤2.5 )的化合物[79]。
建议和注意事项
确保药物筛选库中化合物的分子量在300以下。类先导筛选库对药物的发现至关重要。然而,对商用筛选化合物的近期分析结果表明,药物的分子量中位数为387,平均绝对偏差为78,这说明大部分商用筛选化合物将无法覆盖分子量在300以下的化学空间[80]。这就意味着许多药物筛选的工作可能将无法识别分子量低于300的药物,这部分的药物约占人类药物靶点对的一半。幸运的是,随着人们越来越关注基于片段的药物发现,商用类先导筛选库的数量也越来越多了[81-83]。如果将类先导筛选库作优化用途,它还可以通过表型筛选和基于片段的化学蛋白质组学来识别脂肪细胞分化启动子[84]。类先导药物的发现与“药物化学家的视角”有关,许多药物化学家都讨论过类先导药物的比较分类[85]。也就是说,这项调查表明了药物化学家在评估类先导药物及其分类的时候存在相对的偏见和分化。当药物筛选应用到发现类先导药物时,在筛选的初始阶段可能会存在一种偏见,这种偏见会根据药物分子量过早地把一些类先药物排除掉。
令人欣慰的是,这篇综述讨论的16个例子代表了不同类型的靶标如酶、GPCRs、离子通道和转运蛋白等。此外,最近对未知的口袋和次要的结合口袋进行了研究[86-88],通过变构、诱导配合或其他机制的途径,可能将类先导药物的靶向空间延伸到表征较少的靶标上。
重视分子量中等和分子量受限的药物化学
分子量低于或者等于300的分子通常会被认为是类先导药物,这种化合物需要通过药物化学优化来提供药物,无论是在它药效上的优化还是性能上的优化。然而,本文通过对比单胺类GPCR家族中苗头化合物和优化后化合物的分子量范围,发现分子量﹤300的类先导药物在优化后,它的分子量变化中值为30(平均变化值为50)[89]。这意味着,优化后化合物的分子量与它们的类先导药物的分子量相似。分析发现除了单胺GPCRs外,离子通道和转运蛋白是与低分子量配体相关的蛋白靶标。支持这一观点的是离子通道靶标类的临床候选药物,它们的平均分子质量明显较低。也就是说,针对一些特定的靶标类型,类先导药物可能更适用于临床候选药物的优化工作,因为这些药物需要保持低分子量[90]。此外,识别具有较低表面积的结合口袋也可能为发现类先导药物提供机会[91]。
类先导药物筛选策略的实施是可行的,而类先导药物的药物化学优化工作虽然具有挑战性,但也可能得到预期的结果。通过使用生化分析手段的高浓度的片段筛选已经证明了它的实用性,由于筛选化合物的浓度高,片段筛选能够高灵敏度地鉴定出类先导药物,同时保持可接受的假阳性率[92]。与SPR和NMR基于片段的筛选技术相比,生物化学分析中的片段筛选技术更适合于丝氨酸蛋白酶和金属蛋白酶靶标的鉴定,可以降低蛋白质的需求量[93]。在碎片化过程中保持分子质量较低来引导药物化学研究是很困难的。Johnson和他的同事对2015年-2016年发表的《领先药物化学》的片段分析表明,在55篇类先导药物分子量低于300的文章中,只有6篇产生了分子量低于300的先导化合物[94,95]。我们建议在研究中应用某些药物化学策略,比如构象限制、重原子取代、维持或降低分子量的截断;而不建议应用诸如片段生长并结合到其他的口袋、片段连接或附加增溶基团等策略的应用。如何以及何时使用类先导筛选和药物化学策略还需要进一步的工作来确定。
评估生物学和临床选择
最后,不需要大规模的筛选或广泛的药物化学工作的药物来源不应被忽视。已知药物的代谢物、天然产物的生物学研究以及临床驱动的已知药物的老药新用都可以产生新药批准。
结论
分子量在300以下的化合物是药物研究硕果累累的一个领域,近期该领域出现了许多获批药物。这些新开发的药物中有许多是与已知药物或天然底物类似的药物,对这些化合物的优化的重点是选择低分子量的苗头化合物,并在优化过程中保持低分子量。类先导药物的优点可能还包括减少毒性和改善吸收和清除率。然而,低分子量类先导药物的识别可能会受到合适的低分子量筛选库的实用性的限制。为了提高类先导药物的发现率,我们建议增加对筛选文库中类先导筛选库的关注。在所有的研究案例中都从类先导药物中产生临床候选药物和是不可能的,但历史和最近的药物批准表明,进一步研究这一领域的时机成熟了。
文献
1. Walters, W. P.; Green, J.; Weiss, J. R.; Murcko, M. A. What Do Medicinal Chemists Actually Make? A 50-Year Retrospective. J. Med. Chem. 2011, 54 (19), 6405−6416.
2. Lipinski, C. A. Lead- and Drug-Like Compounds: The Rule-of Five Revolution. Drug Discovery Today: Technol. 2004, 1 (4), 337−341.
3. Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for Fragment-Based Lead Discovery? Drug Discovery Today 2003, 8 (19), 876−877.
4. Quinton, J.; Charruault, L.; Nevers, M. C.; Volland, H.; Dognon, J. P.; Creminon, C.; Taran, F. Toward the Limits of Sandwich Immunoassay of Very Low Molecular Weight Molecules. Anal. Chem. 2010, 82 (6), 2536−2540.
5. The Top 200 of 2017. http://clincalc.com/DrugStats/Top200Drugs.aspx (accessed November 17, 2017).
6. Flick, A. C.; Ding, H. X.; Leverett, C. A.; Kyne, R. E., Jr.; Liu, K. K.; Fink, S. J.; O’Donnell, C. J. Synthetic Approaches to the 2014 New Drugs. Bioorg. Med. Chem. 2016, 24 (9), 1937−1980.
7. Elewski, B. E.; Aly, R.; Baldwin, S. L.; Gonzalez Soto, R. F.; Rich, P.; Weisfeld, M.; Wiltz, H.; Zane, L. T.; Pollak, R. Efficacy and Safety of Tavaborole Topical Solution, 5%, a Novel Boron-Based Antifungal Agent, for the Treatment of Toenail Onychomycosis: Results from 2 Randomized Phase-III Studies. J. Am. Acad. Dermatol. 2015, 73 (1), 62−69.
8. Benkovic, S. J.; Baker, S. J.; Alley, M. R.; Woo, Y. H.; Zhang, Y. K.; Akama, T.; Mao, W.; Baboval, J.; Rajagopalan, P. T.; Wall, M.; Kahng, L. S.; Tavassoli, A.; Shapiro, L. Identification of Borinic Esters as Inhibitors of Bacterial Cell Growth and Bacterial Methyltransferases, Ccrm and Menh. J. Med. Chem. 2005, 48 (23), 7468−7476.
9. Baker, S. J.; Zhang, Y. K.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M. R.; Sanders, V.; Plattner, J. J. Discovery of a New Boron-Containing Antifungal Agent, 5-Fluoro-1,3-Dihydro-1-Hydroxy-2,1- Benzoxaborole (AN2690), for the Potential Treatment of Onychomycosis. J. Med. Chem. 2006, 49 (15), 4447−4450.
10. Paton, D. M. Crisaborole: Phosphodiesterase Inhibitor for Treatment of Atopic Dermatitis. Drugs Today 2017, 53 (4), 239−245.
11. Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of Premature Stop Codon Readthrough in the Cftr Gene by Ataluren (PTC124) Derivatives. Eur. J. Med. Chem. 2015, 101, 236−244.
12. Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a Rationale for the Ptc124 (Ataluren) Promoted Readthrough of Premature Stop Codons: A Computational Approach and GFP-Reporter Cell-Based Assay. Mol. Pharmaceutics 2014, 11 (3), 653−664.
13. Karp, G. M.; Hwang, S.; Chen, G.; Almstead, N. G.; Moon, Y.-C. 1,2,4-Oxadiazole Benzoic Acid Compounds and Their Use for Nonsense Suppression and the Treatment of Disease. US6992096B2, 2004.
14. Bang-Andersen, B.; Ruhland, T.; Jorgensen, M.; Smith, G.; Frederiksen, K.; Jensen, K. G.; Zhong, H.; Nielsen, S. M.; Hogg, S.; Mork, A.; Stensbol, T. B. Discovery of 1-[2-(2,4-Dimethylphenylsulfanyl)Phenyl]Piperazine (Lu AA21004): A Novel Multimodal Compound for the Treatment of Major Depressive Disorder. J. Med. Chem. 2011, 54 (9), 3206−3221.
15. Edaravone Acute Infarction Study Group. Effect of a Novel Free Radical Scavenger, Edaravone (MCI-186), on Acute Brain Infarction. Randomized, Placebo-Controlled, Double-Blind Study at Multicenters. Cerebrovasc. Dis. 2003, 15 (3), 222−229.
16. Abe, K.; Yuki, S.; Kogure, K. Strong Attenuation of Ischemic and Postischemic Brain Edema in Rats by a Novel Free Radical Scavenger. Stroke 1988, 19 (4), 480−485.
17. Brune, K. The Early History of Non-Opioid Analgesics. Acute Pain 1997, 1 (1), 33−40.
18. Hardeland, R. Investigational Melatonin Receptor Agonists. Expert Opin. Invest. Drugs 2010, 19 (6), 747−764.
19. Vachharajani, N. N.; Yeleswaram, K.; Boulton, D. W. Preclinical Pharmacoki-
netics and Metabolism of BMS-214778, a Novel Melatonin Receptor Agonist. J. Pharm. Sci. 2003, 92 (4), 760−772.
20. Halford, J. C.; Harrold, J. A.; Lawton, C. L.; Blundell, J. E.Serotonin (5-HT) Drugs: Effects on Appetite Expression and Use for the Treatment of Obesity. Curr. Drug Targets 2005, 6 (2), 201−213.
21. Smith, B. M.; Smith, J. M.; Tsai, J. H.; Schultz, J. A.; Gilson, C. A.; Estrada, S. A.; Chen, R. R.; Park, D. M.; Prieto, E. B.; Gallardo, C. S.; Sengupta, D.; Dosa, P. I.; Covel, J. A.; Ren, A.; Webb, R. R.; Beeley, N. R.; Martin, M.; Morgan, M.; Espitia, S.; Saldana, H. R.; Bjenning, C.; Whelan, K. T.; Grottick, A. J.; Menzaghi, F.; Thomsen, W. J. Discovery and Structure-Activity Relationship of (1R)-8-Chloro-2,3,4,5-Tetrahydro-1-Methyl-1h-3-Benzazepine (Lorcaserin), a Selective Serotonin 5-HT2c Receptor Agonist for the Treatment of Obesity. J. Med. Chem. 2008, 51 (2), 305−313.
22. Brashier, D. B.; Sharma, A. K.; Dahiya, N.; Singh, S. K.; Khadka, A. Lorcaserin: A Novel Antiobesity Drug. J. Pharmacol. Pharmacother. 2014, 5 (2), 175−178.
23. Rogawski, M. A. Brivaracetam: A Rational Drug Discovery Success Story. Br. J. Pharmacol. 2008, 154 (8), 1555−1557.
24. Klitgaard, H.;Matagne, A.;Nicolas, J. M.;Gillard, M.;Lamberty, Y.;De Ryck, M.; Kaminski, R. M.; Leclercq, K.; Niespodziany, I.; Wolff, C.; Wood, M.; Hannestad, J.; Kervyn, S.; Kenda, B. Brivaracetam: Rationale for Discovery and Preclinical Profile of a Selective SV2A Ligand for Epilepsy Treatment. Epilepsia 2016, 57(4), 538-548.
25. Russo, E.; Citraro, R.; Mula, M. The Preclinical Discovery and Development of Brivaracetam for the Treatment of Focal Epilepsy. Expert Opin. Drug Discovery 2017, 12 (11), 1169−1178.
26. Weinberg, B. A.; Marshall, J. L.; Salem, M. E. Trifluridine/Tipiracil and Regorafenib: New Weapons in the War against Metastatic Colorectal Cancer. Clin Adv. Hematol. Oncol. 2016, 14 (8), 630−638.
27. Fukushima, M.; Suzuki, N.; Emura, T.; Yano, S.; Kazuno, H.; Tada, Y.;Yamada, Y.; Asao, T. Structure and Activity of Specific Inhibitors of Thymidine Phosphory-lase to Potentiate the Function of Antitumor 2’-Deoxyribonucleosides. Biochem. Pharmacol. 2000, 59 (10), 1227−1236.
28. Peters, G. J.;De Bruin, M.;Fukushima, M.;Van Triest, B.;Hoekman, K.;Pinedo, H.M.;Ackland, S. P. Thymidine Phosphorylase in Angiogenesis and Drug Resistance. Homology with Platelet-Derived Endothelial Cell Growth Factor. Adv. Exp. Med. Biol.2000, 486, 291−294.
29. Takao, S.; Akiyama, S. I.; Nakajo, A.; Yoh, H.; Kitazono, M.; Natsugoe, S.; Miyadera, K.; Fukushima, M.; Yamada, Y.; Aikou, T. Suppression of Metastasis by Thymidine Phosphorylase Inhibitor. Cancer Res. 2000, 60 (19), 5345−5348.
30. Cleary, J. M.; Rosen, L. S.; Yoshida, K.; Rasco, D.; Shapiro, G. I.; Sun, W. A Phase 1 Study of the Pharmacokinetics of Nucleoside Analog Trifluridine and Thy-midine Phosphorylase Inhibitor Tipiracil (Components of TAS-102) Vs Trifluridine Alone. Invest. New Drugs 2017, 35 (2), 189−197.
31. Rios-Tamayo, R.; Martin-Garcia, A.; Alarcon-Payer, C.; Sanchez-Rodriguez, D.; de la Guardia, A.; Garcia Collado, C. G.; Jimenez Morales, A.; Jurado Chacon, M.; Cabeza Barrera, J. Pomalidomide in the Treatment of Multiple Myeloma: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2017, 11, 2399−2408.
32. Muller, G. W.; Corral, L. G.; Shire, M. G.; Wang, H.; Moreira, A.; Kaplan, G.; Stirling, D. I. Structural Modifications of Thalidomide Produce Analogs with Enhanced Tumor Necrosis Factor Inhibitory Activity. J. Med. Chem. 1996, 39 (17), 3238−3240.
33. D Amato, R. J.; Lentzsch, S.; Anderson, K. C.; Rogers, M. S. Mechanism of Action of Thalidomide and 3-Aminothalidomide in Multiple Myeloma. Semin. Oncol. 2001, 28 (6), 597−601.
34. Schwartz, J. C. The Histamine H3 Receptor: From Discovery to Clinical Trials with Pitolisant. Br. J. Pharmacol. 2011, 163 (4), 713−721.
35. Ropper, A. H. The “Poison Chair” Treatment for Multiple Sclerosis. N. Engl. J. Med. 2012, 367 (12), 1149−1150.
36. Held, K. D.; Epp, E. R.; Clark, E. P.; Biaglow, J. E. Effect of Dimethyl Fumarate on the Radiation Sensitivity of Mammalian Cells in Vitro. Radiat. Res. 1988, 115 (3), 495−502.
37. Schimrigk, S.; Brune, N.; Hellwig, K.; Lukas, C.; Bellenberg, B.; Rieks, M.; Hoffmann, V.; Pohlau, D.; Przuntek, H. Oral Fumaric Acid Esters for the Treatment of Active Multiple Sclerosis: An Open-Label, Baseline-Controlled Pilot Study. Eur. J. Neurol 2006, 13 (6), 604−610.
38. Gold, R.; Kappos, L.; Arnold, D. L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M. T.; Yang, M.; Sheikh, S. I.; Dawson, K. T. Investigators, D. S. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367 (12), 1098−1107.
39. McGuire, V. A.; Ruiz-Zorrilla Diez, T.; Emmerich, C. H.; Strickson, S.; Ritorto, M. S.; Sutavani, R. V.; Weiss, A.; Houslay, K. F.; Knebel, A.; Meakin, P. J.; Phair, I. R.; Ashford, M. L.; Trost, M.; Arthur, J. S. Dimethyl Fumarate Blocks Pro-Inflammatory Cytokine Production Via Inhibition of TLR Induced M1 and K63 Ubiquitin Chain Formation. Sci. Rep. 2016, 6, 31159.
40. Susitaival, P.; Winhoven, S. M.; Williams, J.; Lammintausta, K.; Hasan, T.; Beck, M. H.; Gruvberger, B.; Zimerson, E.; Bruze, M. An Outbreak of Furniture Related Dermatitis (“Sofa Dermatitis”) in Finland and the UK: History and Clinical Cases. J. Eur. Acad. Dermatol. Venereol. 2010, 24 (4), 486−489.
41. Miyake, Y.; Ebata, M. Galactostatin, a New Beta-Galactosidase Inhibitor from Streptomyces Lydicus. J. Antibiot. 1987, 40 (1), 122−123.
42. Sanchez-Fernandez, E. M.; Garcia Fernandez, J. M.; Mellet, C. O. Glycomimetic-Based Pharmacological Chaperones for Lysosomal Storage Disorders: Lessons from Gaucher, GM1-Gangliosidosis and Fabry Diseases. Chem. Commun. (Cambridge, U. K.) 2016, 52 (32), 5497−5515.
43. Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S. A.; Desnick, R. J.; Benjamin, E. R.; Lockhart, D. J.; Valenzano, K. J. The Pharmacological Chaperone 1-Deoxygalactonojirimycin Reduces Tissue Globotriaosylceramide Levels in a Mouse Model of Fabry Disease. Mol. Ther. 2010, 18 (1), 23−33.
44. Benjamin, E. R.; Flanagan, J. J.; Schilling, A.; Chang, H. H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R. J.; Lockhart, D. J.; Valenzano, K. J. The Pharmacological Chaperone 1-Deoxygalactonojirimycin Increases Alpha-Galactosidase a Levels in Fabry Patient Cell Lines. J. Inherited Metab. Dis. 2009, 32 (3), 424-440.
45. Abian, O.; Alfonso, P.; Velazquez-Campoy, A.; Giraldo, P.; Pocovi, M.;Sancho, J. Therapeutic Strategies for Gaucher Disease: Miglustat (NB-DNJ) as a Pharmaco-logical Chaperone for Glucocerebrosidase and the Different Thermostability of Ve-laglucerase Alfa and Imiglucerase. Mol. Pharmaceutics 2011, 8 (6), 2390−2397.
46. Schiffmann, R. Agalsidase Treatment for Fabry Disease: Uses and Rivalries. Genet. Med. 2010, 12 (11), 684−685.
47. Schiffmann, R.; Martin, R. A.; Reimschisel, T.; Johnson, K.; Castaneda, V.; Lien, Y. H.; Pastores, G. M.; Kampmann, C.; Ries, M.; Clarke, J. T. Four-Year Prospective Clinical Trial of Agalsidase Alfa in Children with Fabry Disease. J. Pediatr. 2010, 156 (5), 832−837.
48. Benjamin, E. R.; Della Valle, M. C.; Wu, X.; Katz, E.; Pruthi, F.; P.; Giugliani, R.; Hughes, D.; Schiffmann, R.; Wilcox, W. R.; Desnick, R. J.; Kirk, J.; Barth, J.; Barlow, C.; Valenzano, K. J.; Castelli, J.; Lockhart, D. J. The Validation of Pharma-cogenetics for the Identification of Fabry Patients to Be Treated with Migalastat. Genet. Med. 2017, 19 (4), 430−438.
49. Antoniu, S. A. Pirfenidone for the Treatment of Idiopathic Pulmonary Fibrosis. Expert Opin. Invest. Drugs 2006, 15 (7), 823−828.
50. Genentech Mission Critical. https://www.gene.com/stories/mission-critical (accessed November 17, 2017).
51. Nathan, S. D.; Albera, C.; Bradford, W. Z.; Costabel, U.; Glaspole, I.; Glassberg, M. K.; Kardatzke, D. R.; Daigl, M.; Kirchgaessler, K.-U.; Lancaster, L. H.; Lederer, D. J.; Pereira, C. A.; Swigris, J. J.; Valeyre, D.; Noble, P. W. Effect of Pirfenidone on Mortality: Pooled Analyses and Meta-Analyses of Clinical Trials in Idiopathic Pulmonary Fibrosis. Lancet Respir. Med. 2017, 5 (1), 33−41.
52. Noble, P. W.; Albera, C.; Bradford, W. Z.; Costabel, U.; Glassberg, M. K.; Kardatzke, D.; King, T. E.; Lancaster, L.; Sahn, S. A.; Szwarcberg, J.; Valeyre, D.; du Bois, R. M. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis(Capacity): Two Randomised Trials. Lancet 2011, 377 (9779), 1760−1769.
53. Macias-Barragan, J.; Sandoval-Rodriguez, A.; Navarro-Partida, J.; Armendariz-Borunda, J. The Multifaceted Role of Pirfenidone and Its Novel Targets. Fibrog. Tissue Repair 2010, 3, 16.
54. Nanthakumar, C. B.; Hatley, R. J.; Lemma, S.; Gauldie, J.; Marshall, R. P.; Macdonald, S. J. Dissecting Fibrosis: Therapeutic Insights from the Small-Molecule Toolbox. Nat. Rev. Drug Discovery 2015, 14 (10), 693−720.
55. Bertolini, G.; Aquino, M.; Biffi, M.; d’Atri, G.; Di Pierro, F.; Ferrario, F.; Mascagni, P.; Somenzi, F.; Zaliani, A.; Leoni, F. A New Rational Hypothesis for the Pharmacophore of the Active Metabolite of Leflunomide, a Potent ImmunoSuppre-ssive Drug. J. Med. Chem. 1997, 40 (13), 2011−2016.
56. Munier-Lehmann, H.; Vidalain, P. O.; Tangy, F.; Janin, Y. L. On Dihydrooro-tate Dehydrogenases and Their Inhibitors and Uses. J. Med. Chem. 2013, 56 (8), 3148−3167.
57. Benes, J.; Parada, A.; Figueiredo, A. A.; Alves, P. C.; Freitas, A. P.; Learmonth, D. A.; Cunha, R. A.; Garrett, J.; Soares-da-Silva, P. Anticonvulsant and Sodium Channel-Blocking Properties of Novel 10,11-Dihydro-5h-Dibenz[b,f] Azepine-5-Carboxamide Derivatives. J. Med. Chem. 1999, 42 (14), 2582−2587.
58. Droxidopa, a prodrug, has six hydrogen bond donors, but the active entity, the neurotransmitter, norepinephrine has fifive and therefore is not an outlier to the rule-of-fifive concept.
59. Veber, D. F.; Johnson, S. R.; Cheng, H. Y.; Smith, B. R.; Ward, K. W.; Kopple, K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45 (12), 2615−2623.
60. Leeson, P. D. Molecular Inflation, Attrition and the Rule of Five. Adv. Drug Delivery Rev. 2016, 101, 22−33.
61. Hopkins, A. L.; Groom, C. R.; Alex, A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discovery Today 2004, 9 (10), 430−431.
62. Hopkins, A. L.; Keseru, G. M.; Leeson, P. D.; Rees, D. C.; Reynolds, C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105−121.
63. Egan, W. J.; Merz, K. M., Jr.; Baldwin, J. J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43 (21), 3867−3877.
64. Chmielewska, A.; Lamparczyk, H. Mass Versus Molar Doses, Similarities and Differences. Pharmazie 2008, 63 (11), 843−848.
65. Varma,M.V.;Gardner, I.;Steyn,S.J.;Nkansah,P.;Rotter,C.J.;Whitney-Pickett, C.; Zhang, H.; Di, L.; Cram, M.; Fenner, K. S.; El Kattan, A. F. Ph-Dependent Solubil-ity and Permeability Criteria for Provisional Biopharmaceutics Classification (BCS and BDDCS) in Early Drug Discovery. Mol. Pharmaceutics 2012, 9 (5), 1199−1212.
66. Huuskonen, J.; Livingstone, D. J.; Manallack, D. T. Prediction of Drug Solubi-lity from Molecular Structure Using a Drug-Like Training Set. SAR QSAR Environ. Res. 2008, 19 (3−4), 191−212.
67. Yazdanian, M.; Glynn, S. L.; Wright, J. L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15 (9), 1490−1494.
68. Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14 (5), 568−571.
69. Artursson, P.; Ungell, A. L.; Lofroth, J. E. Selective Paracellular Permeability in Two Models of Intestinal Absorption: Cultured Monolayers of Human Intestinal Epithelial Cells and Rat Intestinal Segments. Pharm. Res. 1993, 10 (8), 1123−1129.
70. Johnson, T. W.; Dress, K. R.; Edwards, M. Using the Golden Triangle to Optimize Clearance and Oral Absorption. Bioorg. Med. Chem. Lett. 2009, 19 (19), 5560−5564.
71. Rankovic, Z. CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain. J. Med. Chem. 2017, 60 (14), 5943−5954.
72. Smith, D. A.; Beaumont, K.; Maurer, T. S.; Di, L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58 (15), 5691−5698.
73. Smith, D. A.; Beaumont, K.; Maurer, T. S.; Di, L. Relevance of Half-Life in Drug Design. J. Med. Chem. 2018, 61 (10), 4273−4282.
74. Sharifi, M.; Ghafourian, T. Estimation of Biliary Excretion of Foreign Compounds Using Properties of Molecular Structure. AAPS J. 2014, 16 (1), 65−78.
75. Fleck, C.; Braunlich, H. Factors Determining the Relationship between Renal and Hepatic Excretion of Xenobiotics. Arzneimittelforschung 1990, 40 (8), 942-946.
76. Varma, M. V.; Lai, Y.; El-Kattan, A. F. Molecular Properties Associated with Transporter-Mediated Drug Disposition. Adv. Drug Delivery Rev. 2017, 116, 92-99.
77. Meanwell, N. A. Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space. Chem. Res. Toxicol. 2016, 29 (4), 564−616.
78. Struck, S.; Schmidt, U.; Gruening, B.; Jaeger, I. S.; Hossbach, J.; Preissner, R. Toxicity Versus Potency: Elucidation of Toxicity Properties Discriminating between Toxins, Drugs, and Natural Compounds. Genome Inform 2008, 20, 231−242.
79. Price, D.; Lee, K. Unpublished results.
80. Zuegg, J.; Cooper, M. A. Drug-likeness and Increased Hydrophobicity of Commercially Available Compound Libraries for Drug Screening. Curr. Top. Med. Chem. 2012, 12 (14), 1500−1513.
81. de Kloe, G. E.; Bailey, D.; Leurs, R.; de Esch, I. J. Transforming Fragments into Candidates: Small Becomes Big in Medicinal Chemistry. Drug Discovery Today 2009, 14 (13−14), 630−646.
82. Chessari, G.; Woodhead, A. J. From Fragment to Clinical Candidate-aHistorical Perspective. Drug Discovery Today 2009, 14 (13−14), 668−675.
83. Keseru, G. M.; Erlanson, D. A.; Ferenczy, G. G.; Hann, M. M.; Murray, C. W.; Pickett, S. D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59 (18), 8189−8206.
84. Parker, C. G.; Galmozzi, A.; Wang, Y.; Correia, B. E.; Sasaki, K.; Joslyn, C. M.; Kim, A. S.; Cavallaro, C. L.; Lawrence, R. M.; Johnson, S. R.; Narvaiza, I.; Saez, E.; Cravatt, B. F. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527−541.
85. Lajiness, M. S.; Maggiora, G. M.; Shanmugasundaram, V. Assessment of the Consistency of Medicinal Chemists in Reviewing Sets of Compounds. J. Med. Chem. 2004, 47 (20), 4891−4896.
86. Oleinikovas, V.; Saladino, G.; Cossins, B. P.; Gervasio, F. L. Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations. J. Am. Chem. Soc. 2016, 138 (43), 14257−14263.
87. Ludlow, R. F.; Verdonk, M. L.; Saini, H. K.; Tickle, I. J.; Jhoti, H. Detection of Secondary Binding Sites in Proteins Using Fragment Screening. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (52), 15910−15915.
88. Hart, K. M.; Moeder, K. E.; Ho, C. M. W.; Zimmerman, M. I.; Frederick, T. E.; Bowman, G. R. Designing Small Molecules to Target Cryptic Pockets Yields Both Positive and Negative Allosteric Modulators. PLoS One 2017, 12 (6), e0178678.
89. Morphy, R. The Influence of Target Family and Functional Activity on the Physicochemical Properties of Pre-Clinical Compounds. J. Med. Chem. 2006, 49(10), 2969−2978.
90. Waring, M. J.; Arrowsmith, J.; Leach, A. R.; Leeson, P. D.; Mandrell, S.; Owen, R. M.; Pairaudeau, G.; Pennie, W. D.; Pickett, S. D.; Wang, J.; Wallace, O.; Weir, A. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discovery 2015, 14 (7), 475−486.
91. Cheng, A. C.; Coleman, R. G.; Smyth, K. T.; Cao, Q.; Soulard, P.; Caffrey, D. R.; Salzberg, A. C.; Huang, E. S. Structure-Based Maximal Affinity Model Predicts Small-Molecule Druggability. Nat. Biotechnol. 2007, 25 (1), 71−75.
92. Barker,J.; Courtney,S.; Hesterkamp,T.; Ullmann,D.; Whittaker,M. Fragment Screening by Biochemical Assay. Expert Opin.Drug Discovery 2006,1(3),225-236.
93. Boettcher, A.; Ruedisser, S.; Erbel, P.; Vinzenz, D.; Schiering, N.; Hassiepen, U.; Rigollier, P.; Mayr, L. M.; Woelcke, J. Fragment Based Screening by Biochemical Assays: Systematic Feasibility Studies with Trypsin and MMP12. J. Biomol. Screening 2010, 15 (9), 1029−1041.
94. Johnson, C. N.; Erlanson, D. A.; Murray, C. W.; Rees, D. C. Fragment-to-Lead Medicinal Chemistry Publications in 2015. J. Med. Chem. 2017, 60 (1), 89−99.
95. Johnson, C. N.; Erlanson, D. A.; Jahnke, W.; Mortenson, P. N.; Rees, D. C. Fragment-to-Lead Medicinal Chemistry Publications in 2016. J. Med. Chem. 2018, 61 (5), 1774−1784.


