
摘要:在本研究中,以PBDbind数据库(2014版)的2002个蛋白-配体复合物结构作为测试用数据集,评估了10个分子对接软件的采样性能与打分性能。结果表明,GOLD与LeDock显示出最好的采样性能(GOLD:打分最佳的结合模式正确率为59.8%;LeDock:最佳结合模式的正确率为80.8%);AutoDock Vina、GOLD与MOE Dock的打分性能表现最佳,其打分最佳结合模式的rp/rs分别为0.564/0.580、0.500/0.515与0.569/0.589。然而,打分值与实验测得的结合亲合力之间的相关性还相当弱,这说明打分函数不太可靠、通用性也不足。打分性能的评估也表明,同一软件对不同蛋白家族的打分性能差异很大(rp 为0.000~0.800) ,因此不同蛋白家族的对接研究可能需要使用不同的软件。评价结果还发现没有一个软件可以全面优于其它软件。在一个计算平台组合使用几种对接软件或许是提高基于对接虚拟筛选精度的实用方法。
原文:Wang, Z., et al. (2016). “Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power.” Phys Chem Chem Phys 18(18): 12964-12975.
编译:肖高铿
一. 为什么要比较不同软件的性能
分子对接技术(Molecular docking)在药物设计领域广受欢迎,它既可以用来预测结合模式、还可以用来预测结合亲合力。在过去的20年中涌现出了大量的分子对接软件比如AutoDock, AutoDock Vina, LeDock, rDock, UCSF DOCK, LigandFit, GLIDE, GOLD, MOE Dock, Surflex-Dock等等,它们既有商业软件也有学术软件。
采样算法与打分函数是对接软件的最核心部分。前者负责将化合物在蛋白口袋里摆pose,采样算法决定了对接软件的采样性能(sampling power);后者负责对每个pose打分,决定了对接软件的打分性能(scoring power)。采样算法负责产生pose,如果它产生的众多pose里不包含有正确的pose,那么打分函数就没有就机会挑到正确的结合模式,对接就会失败;同样地,如果打分函数不能将正确的pose从采样算法产生的可能pose中通过打分、排序后挑出来,那么对接也会失败。
因为不同对接软件的采样算法与打分函数不同、其性能也大不相同,所以评价、比较这些软件的性能就显得非常重要。性能评价的结果可以揭示每个软件的优缺点,并由此帮助用户合理地选择软件。到目前为止,虽然软件评价、比较的文献汗牛充栋,然而最重要的几个研究都是2011年前的事,在最近5年里类似的研究非常有限。但是近几年对接算法快速发展,不但许多传统的对接软件进行了更新换代,而且还出现了一批新的对接软件。相比之下,现有的对接评价研究过于陈旧、不合时宜。总的来说,即使在过去20年有很多的比较研究,但是针对一个特定靶点使用哪个软件更好依旧难以决定。因此广泛、深入地研究当下流行的对接软件的性能是非常有必要的。
在本研究中,作者比较了10种分子对接软件的预测配体结合模式的性能(采样性能)与预测结合亲和力的性能(打分性能)。10个对接软件中有5个是学术软件,5个是商业软件,这些软件的特点见Table 1。大部分商业软件非常昂贵,因此人们也期望商业软件应用物有所值地比学术软件表现出更好的性能。本研究希望能回答下面几个问题:
- 商业软件是否比学术软件更具优势?
- 更流行的软件是否比不那么流行的软件具有更好的性能?
- 传统的软件是否比最新开发的软件性能更好?
在本研究中有三个软件,AutoDock、GOLD与GLIDE,是1990到2013年文章中最流行的软件。那么流行的软件是否优于其它不么流行的软件呢?
LeDock与rDock是新开发的软件,但它们计算速度与精度还比较好,因此本研究中包含了这两个软件。不像传统软件软件,这些新软件验证性文章比较少因而性能还受质疑,因此有必要这个问题。
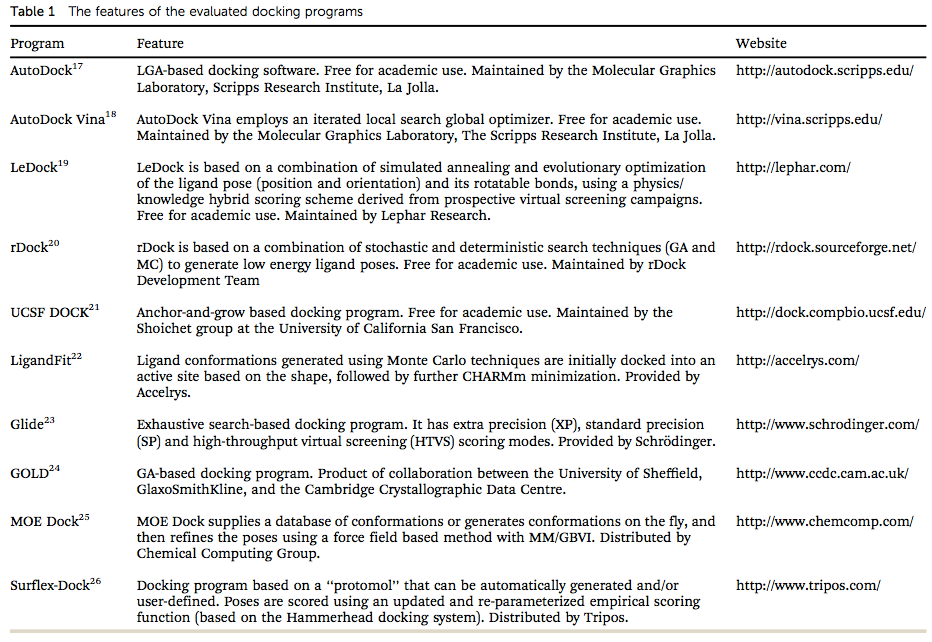
Table 1. 10种分子对接软件及其特点
二. 材料与方法
基准数据集
来自PDBbind优选数据集(refined set)中的2002个蛋白-配体复合物结构。
结构准备
为了考察对接软件采样算法的健壮性,配体使用了三种不同的初始构象:原始构象(original)、旋转构象(rotated)、优化构象(Optimized)。
- 原始构象: 就是从蛋白-配体复合物结构的那个构象
- 旋转构象: 将原始构象绕Z-轴选转180度
- 优化构象: 将旋转构象用OPLS-2005力场优化后的构象
评价方法
- 采样性能(Sample power)
- 打分性能(Scoring power)
与蛋白-配体复合物结构的那个pose比,如果对接计算预测pose的RMSD(root mean square deviation)小于2.0(Å),那么就认为对接计算成功。因为打分最高的那个pose(the top score pose)不一定是对接最佳的那个(the best pose),因此分别考察了打分最高pose的对接成功率与最佳pose的对接成功率。
考察打分函数对化合物结合亲合力的排序能力,用预测的结合亲和力(打分值)与实验值之间的皮尔逊相关系(Pearson’s correlation coefficient,rp)与史皮尔曼等级相关系数(Spearman’s ranking coefficient, rs)来评价。
三. 结果与讨论
3.1 采样性能的评估
Figure 2展示了预测pose与实验pose的RMSD值分布,Figure 3A展示了打分最佳pose与最佳pose的成功率(RMSD小于等于2Å)分布。
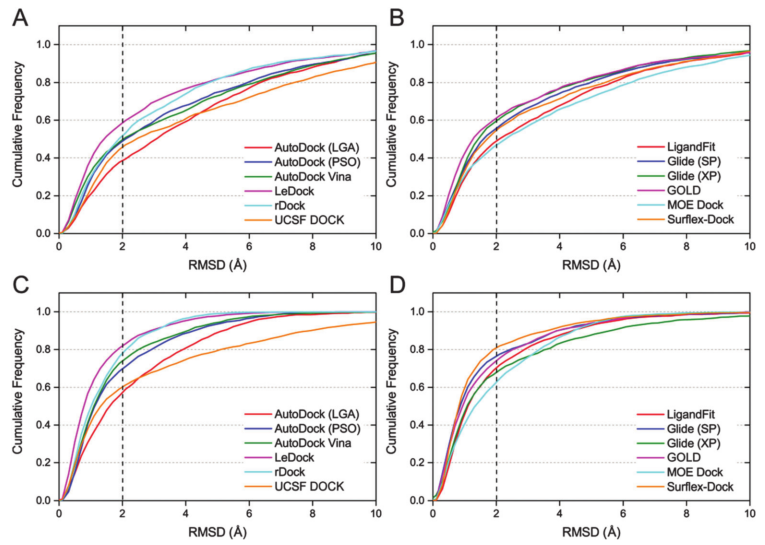
Figure 2. RMSD的累积分布图。A:免费软件用优化构象作为输入,打分最佳的pose;B:商业软件用优化构象作为输入,打分最佳pose;C:免费软件用优化构象作为输入,最佳pose;D:商业软件用优化构象作为输入,最佳pose。
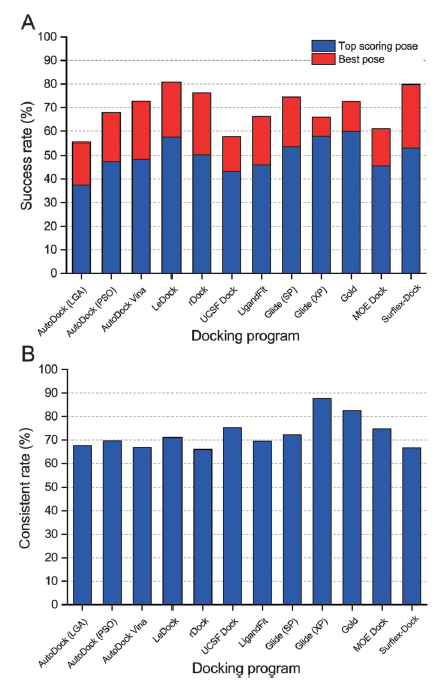
Figure 3.十种对接软件用优化的构象做为初始构象对接计算的成功率(A)与一致率(B), 其中成功的pose其RMSD小于等于2埃
总的来说,如果以优化的构象作为输入进行对接计算,打分最佳pose的成功率为40~60%,最佳pose的成功率为60~80%。以打分最佳的成功率论,学术软件的性能排序为:LeDock(57.4%) > rDock(50.3%) > AutoDock Vina(49.0%) > AutoDock(PSO) (47.3%) > UCSF DOCK(44.0%) > AutoDock(LGA) (37.4%);商业软件的性能排序为:GOLD(59.8%) > Glide XP (57.8%) > Glide SP (53.8%) > Surflex-Dock (53.2%) > LigandFit (46.1%) > MOE Dock (45.6%)。商业软件最佳打分pose与最佳pose的平均成功率分别为54.0%与67.8%;而学术软件的分别为47.4%与68.4%。也就是说,商业软件比起学术软件在结合模式预测上略有优势,但不明显。
免费软件中,LeDock与rDock是比较新的软件,它们的结合模式预测性能令人刮目相看,尤其是LeDock优于大部分商业软件。比较Figure 2B与D会发现Surflex-Dock的最佳pose的成功率为80%,而其打分最佳pose的成功率要远远地低至53.2%。打分最佳pose与最佳pose的预测精度巨大的差距表明Surflex-Dock的排序能力不足、有待提升。另一个出人意料的是GLIDE,它的XP模式居然比SP模式预测成功率低,实际作者在之前的研究也发现XP模式不见得优于SP模式。
在实际的虚拟筛选研究中,通常把打分最佳pose认为是最合理的pose。然而如前所述,我们发现打分最佳的pose通常不是最好的pose,之间差距巨大,这主要是因为打分函数存在缺陷。因此,有必要考察一致率以评估打分最佳pose与最佳pose的一致性。一致率用SRtsp/SRbp来定义,其中SRtsp与SRbp分别是打分最佳pose与最佳pose的成功率。如Figure 3B所示,GLIDE XP与GOLD这两个对接方法的一致率最好,分别为87.7%与82.5%。
3.2 分子柔性对采样性能的影响
在1790个FDA批准的上市药物中,90%药物的柔性键数量小于10。因此考察柔性键数量小于10的配体的对接性能具有更大的价值。结果如Figure 4所示,LeDock、rDock、Glide SP、Glide XP以及GOLD最佳打分pose的成功率较其它软件好,而LeDock、rDock、Glide SP与Surflex-Dock的最佳pose成功率优于其它软件。

Figure 4 不同柔性键数的配体对接成功率热图。(A) 打分最佳pose;(B)最佳pose
3.3 初始构象对采样性能的影响
有文献报道对接计算对配体的初始构象非常敏感,当初始构象与结合构象越相似,预测的结果越好。然而一个健壮的对接软件,其结果不应该因初始构象不同与不同。本研究分别采用了原始构象、旋转构象与优化构象做为初始构象进行结合模式预测,结果如Figure 5所示。AutoDock, AutoDock Vina, LigandFit与GOLD具有相对较大的波动,这说明这几个对接算法对初始构象更敏感。对大部分软件来说,原始构象与旋转构象作为输入文件的结合模式预测精度比优化构象作为输入文件的预测精度更高。在受测试的软件里,LeDock、rDock、UCSF DOCK、Glide、MOE Dock与Surflex-Dock对初始的构象不敏感,也就是说,这几个对接软件的采样算法更稳健。
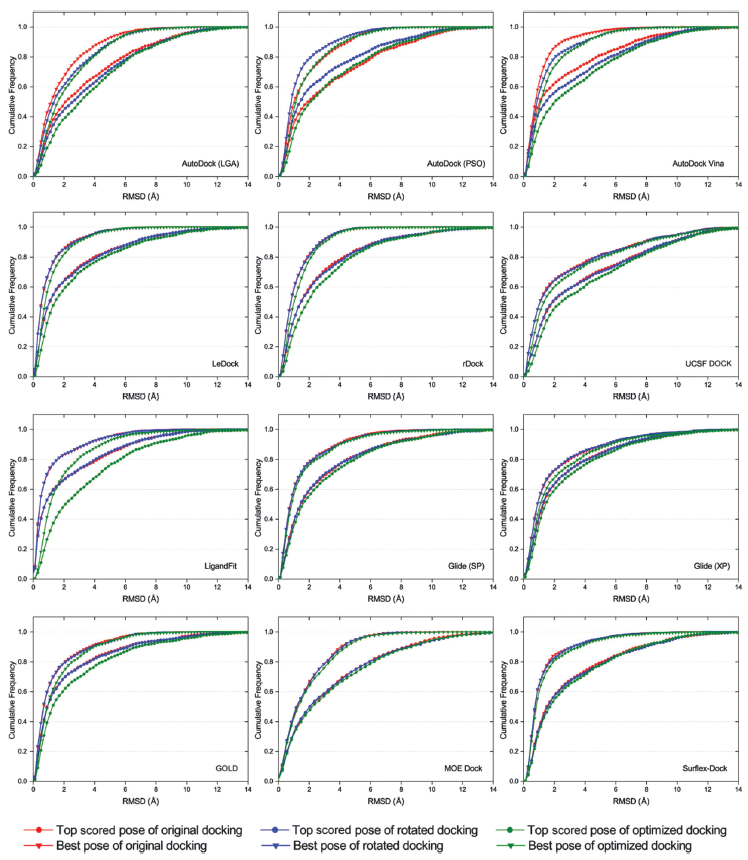
Figure 5 从不同初始构象进行对接计算的打分最佳pose与最佳pose的累积分布图
3.4 打分性能的评估
除了采样性能,对接软件对不同配体结合亲合力的排序(打分性能)是另一个重要的问题,因为对预测生成的构象进行排序是虚拟筛选决定性的步骤。打分性能定义为打分函数对一系列蛋白-配体复合物结合亲合力排序的预测精度。通常一个软件整合有多种打分函数以满足不同计算精度与计算开销。在不同的条件下,打分最佳pose与最佳pose是两种不同的“正确”构象,因此本研究评测了每个软件的这两种打分性能。
Table 3 打分性能测试结果
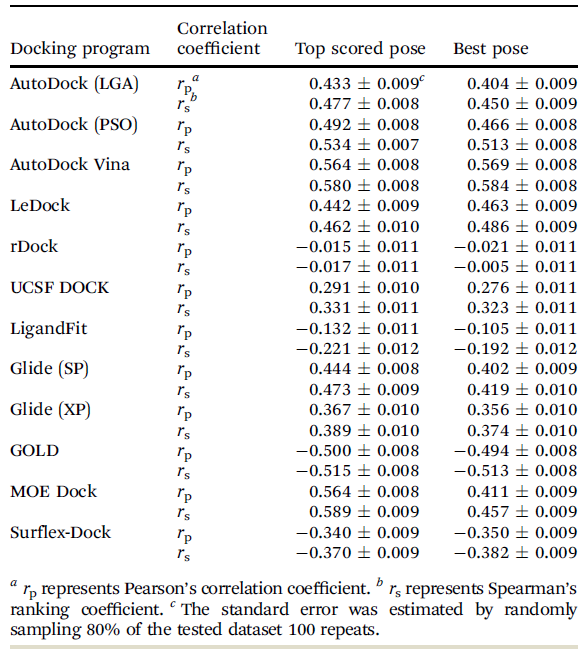
Table 3总结了对接打分值与实验值之间的rp(Pearson correlation coefficient)与rs(Spearman ranking coefficient)。从中可以发现,AutoDock Vina的的打分性能最好,其打分最佳pose与最佳pose的rp(rs)系数分别为0.564(0.580)与0.569(0.584)。排名靠前紧接着是MOE Dock与GOLD,它们的打分最佳pose、最佳pose的rp(rs)分别为0.564(0.589)与0.500(0.511)。出乎意料的是,大部分程序的打分最佳与pose最佳的打分性能没有显著差异,除了MOE Dock之外。总的来说,受试软件的打分性能并不十分令人满意。
根据最佳打分pose的皮尔逊相关系数,学术软件的性能排序如下:AutoDock Vina (0.580) > AutoDock (PSO) (0.534) > LeDock (0.462) > UCSF DOCK (0.331) > rDock (0.017);商业软件的排序如下:MOE Dock (0.589) > GOLD (0.515) > Glide (0.473) > Surflex-Dock (0.370) > LigandFit (0.221)。总的来说,比起学术软件,商业软件在结合亲合力排序上并不占优。此外,可以结合模式性好好的打分函数不一定对结合亲合力排序也好。比如,rDock的采样性能相当好,但是其排序能力相当弱;GOLD的最佳pose采样性能最好,但是它的最佳pose排序性能不是最好的。很显然,没有一个软件可以在采样性能与打分性能两样优于其它软件,因此最好的虚拟筛选解决方案是组合使用几种不同的对接软件。
3.5 打分性能的数据集偏好
如Figure 6所示,同一个软件的打分性能因研究的蛋白家族不同而差异巨大,比如,LeDock对b.47.1.2和b.50.1.2的最佳打分pose的Rp分别为0.698与0.770;但对b.50.1.1与c.94.1.1家族而言,最佳打分pose的Rp分别只有-0.010与0.176。另一方面,不同软件对同一家族的蛋白性能也差异巨大。如Figure 6所示,对b.50.1.2家族而言,AutoDock、rDock与LigandFit的最佳打分pose与最佳pose的Rp都不到0.5;而其它软件如LeDock, Glide (XP), GOLD,与Surflex-Dock的相关性系数大约为0.7或-0.70。这说明选择正确的软件是非常重要的。同时,我们发现全部的软件对b.50.1.1家族的打分性能都不好。
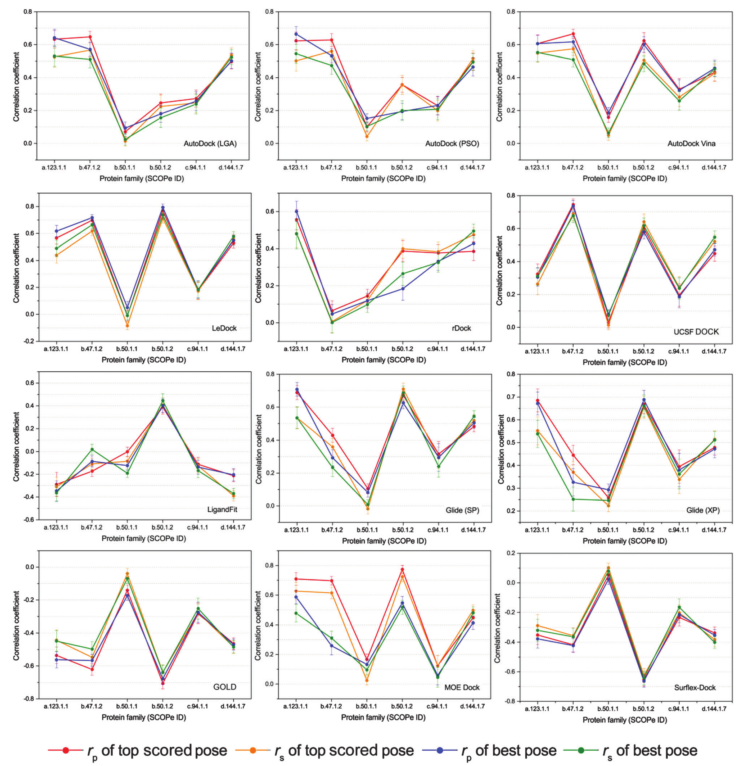
Figure 6 受试软件对至少具有50个成员的几个蛋白家族的打分性能。
四. 结论
在本研究中,以PBDbind数据库(2014版)的2002个蛋白-配体复合物结构作为测试用数据集,评估了10个分子对接软件的采样性能与打分性能。结果表明,GOLD与LeDock显示出最好的采样性能(GOLD:打分最佳的结合模式正确率为59.8%;LeDock:最佳结合模式的正确率为80.8%);AutoDock Vina、GOLD与MOE Dock的打分性能表现最佳,其打分最佳结合模式的rp/rs分别为0.564/0.580、0.500/0.515与0.569/0.589。然而,打分值与实验测得的结合亲合力之间的相关性还相当弱,这说明打分函数不太可靠、通用性也不足。打分性能的评估也表明,同一软件对不同蛋白家族的打分性能差异很大(rp 为0.000~0.800) ,因此不同蛋白家族的对接研究可能需要使用不同的软件。评价结果还发现没有一个软件可以全面优于其它软件。在一个计算平台组合使用几种对接软件或许是提高基于对接虚拟筛选精度的实用方法。


