摘要:在本文中,我们用QPCT/SEN177的共晶结构PDB 6GBX作为QPCTL/SEN177结合模型的代理,回溯性地重现了Yu等人4QPCTL抑制剂的优化过程。首先,发现在PDB 6GBX解释的结构中,SEN177存在两种截然不同的结合模式:Chain A与Chain B。用apo-GIST对结合口袋进行水分析,初步认为Chain B结合模式是正确的,并可以解释现有的SAR。进一步的holo-GIST分析、MMGBSA结合自由计算以及QM相互作用分析,均支持Chain B为正确结合模式,而Chain A的结合模式是不正确的。有了正确的结合模式,然后根据Unhappy 水规划了Spark水分子替换策略对SEN177的吡啶母核进行生物等排体替换实验,重现了文献验证过的QP5020设计。这证明了我们的方法能够替换Unhappy桥连水分子并正确的识别出4-腈基苯片段做为吡啶母核的替换结构。最后,还对三氮唑片段进行了Spark生物等排体替换实验,打分最高排名第3的N-甲基咪唑具有最佳的Zn结合几何模式,并被文献报道的QP5038所验证。Spark还命中尚未有文献报道的新化学骨架类型,有望发展出新的系列化合物。

肖高铿/广州市墨灵格信息科技有限公司
徐锡明/中国海洋大学
2024-02-01:第一次发布。
2024-02-19:补充了Zn离子结合片段三氮唑的生物等排体替换。
2024-06-03:用水合自由能值来代替网格自由能图。
2024-06-10:用GCNCMC代替3D-RISM对水进行增强采样,重新计算水合自由能,并更新了附件。
前言
谷氨酰胺酰肽环转移酶样蛋白(Glutaminyl-peptide cyclotransferase like protein,QPCTL或isoQC)是谷氨酰胺肽环化转移酶(QPCT)的同工酶,催化靶蛋白(如CCL2、CCL7和CX3CL1)上的N-末端谷氨酰胺和谷氨酸残基的环化,形成焦谷氨酸残基。Logtenberg等人1与Wu等人2的研究表明,QPCTL介导了CD47焦谷氨酸的形成,对CD47/SIRPα结合起到至关重要的作用,是调控CD47/SIRPα相互作用的关键因子,并起到“不要吃我(Don’t eat me)”信号的作用。还发现QPCTL缺失显著增强巨噬细胞介导的肿瘤细胞吞噬作用1,2。此外,抑制QPCTL可以通过重塑髓系细胞的浸润来增强PD-1阻断的功效3。这些研究表明,QPCTL是一个有吸引力癌症治疗靶标3。
近几十年来,人们以将靶向QPCTL的小分子抑制剂开发用于阿尔茨海默氏病的治疗,其中一些抑制剂,包括SEN177(图1),也表现出对QPCTL的抑制活性。针对肿瘤免疫治疗的QPCTL抑制剂的研究尚处于早期阶段,因此新型高效的QPCTL抑制剂有待开发。
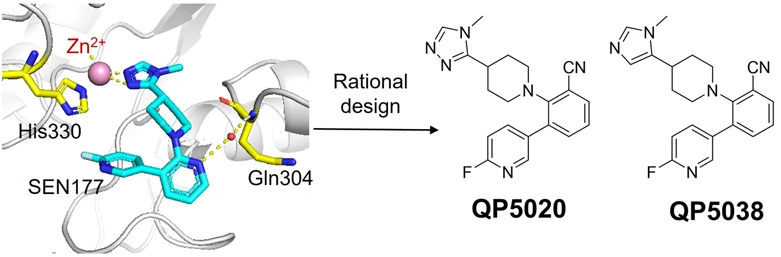
图1. 以SEN177(青蓝色化合物)与QPCT共晶结合模型(PDB 6GBX)为起点设计的QPCTL抑制剂QP5020(化合物4)与QP5038(化合物38)。图片来源于Yu等人的文章4。
最近,Yu等人4报道了利用基于结构的方法以SEN177为起点设计了一系列QPCTL抑制剂。虽然尚未有SEN177与QPCTL的共晶结构,但是QPCT与QPCTL活性位点之间存在高度保守的结构,因此假设SEN177与QPCTL的结合方式与QPCT相似。根据SEN177和QPCT结合模型(PDB 6GBX),一个值得注意的特征是吡啶母核的氮原子与Gln304的骨架NH经由结构水分子介导形成氢键相互作用,如图1所示。为了提高结合亲和力,将SEN177吡啶母核中的氮原子替换为腈基,以产生含苯甲腈的化合物4(QP5020,见图1),因为模拟或置换结合位点水分子是一种行之有效的策略。作者评估了QP5020对QPCTL的抑制活性,结果表明QP5020的活性是SEN177的8.7倍,对QPCTL的IC50 = 15.0±5.5 nM。在进一步对Zn离子结合片段三氮唑优化之后,得到了化合物28(QP5038,见图1),对QPCTL的IC50 = 3.8 ± 0.7 nM。
在2021年8月份的时候,有幸与中国海洋大学的徐锡明博士一起探讨QPCTL/QPCT抑制剂的设计,其中一个系列化合物的设计策略也是用腈基替换PDB 6GBX介导配体与蛋白氢键网络的结合水HOH558,并设计了与QP5020同样的化合物,参见与徐博士的微信聊天截图5。可惜缘分不够,在后续对不同系列进行的优先性排序中,QP5020并没有进入下一轮的合成与活性测试,最终失之交臂。本文将重现这个设计过程并分享对SEN177结合模式的理解。
在本文中,以化合物SEN177以及桥连的水分子HOH558为起始分子用SPARK6进行片段替换,目的是验证我们的方法能否替换桥连水分子并正确的识别出苯甲腈片段做为吡啶母核的替换结构。本文的另一个目的是,用Flare7的GIST分析结合位点的水能量特征,并结合分子对接与量子化学计算认为PDB 6GBX共晶结构中A链的SEN177结合模式可能是错误的,认为B、C链的SEN177结合模式才是正确的,并用Yu等人4公开的系列化合物(表1)来验证我们的假设。
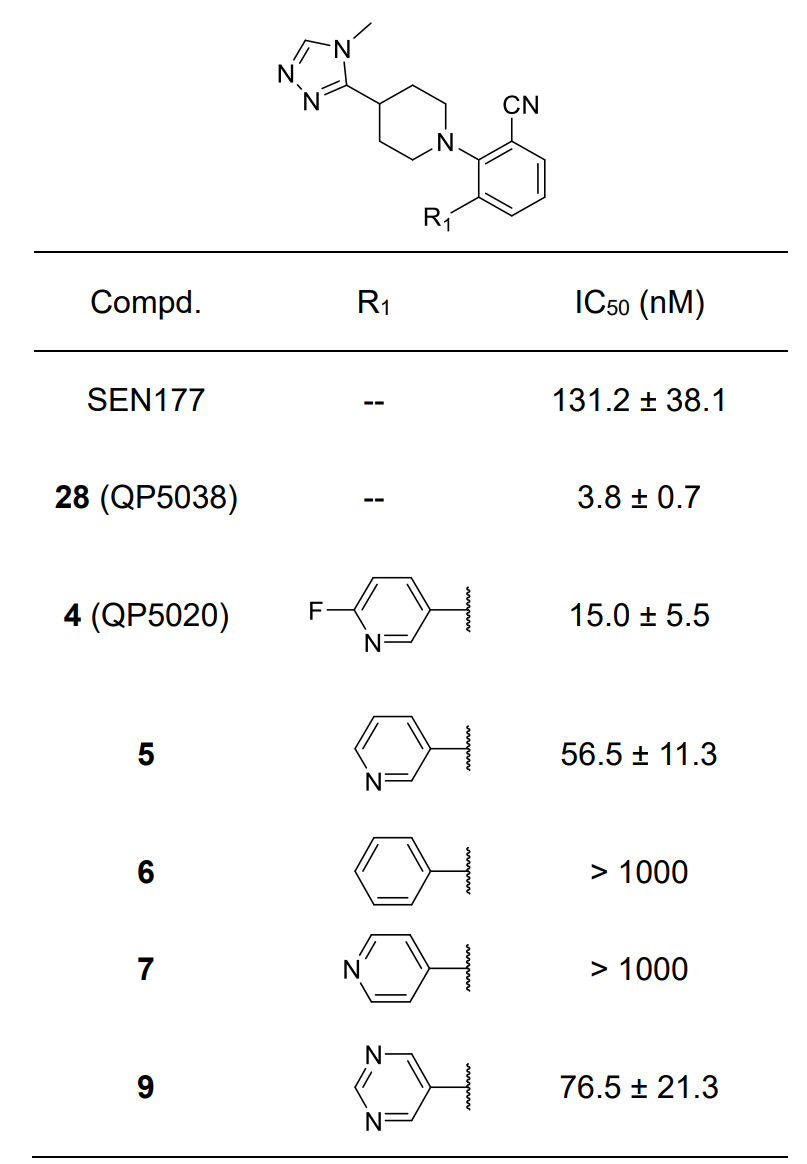
表1. Yu等人4公开的部分QCPTL抑制剂及其体外活性。
最后,还用QM计算方法发现Zn离子结合片段的优化机会,并用QM计算结果指导Spark对Zn结合片段进行骨架跃迁,重现Yu等人4对化合物28的设计。
结果与讨论
有待确认的结合模式与关键的结合水HOH558与HOH581
对PDB结构6GBX的Chain A、B和C进行了分析,发现配体SEN177在不同亚基中的结合模式存在显著差异。如图3所示,在Chain A中,SEN177的末端氟取代吡啶基团的氮原子与水分子HOH581的距离较远,未形成氢键;包括最近解释的另一个SEN177与QCPT的共晶结构PDB 9FXI,也是与Chain A同样的结合模式。然而,在Chain B和Chain C中,SEN177的末端氟取代吡啶基团发生了构象翻转,其氮原子靠近HOH581(即HOH574)并建立了氢键相互作用。
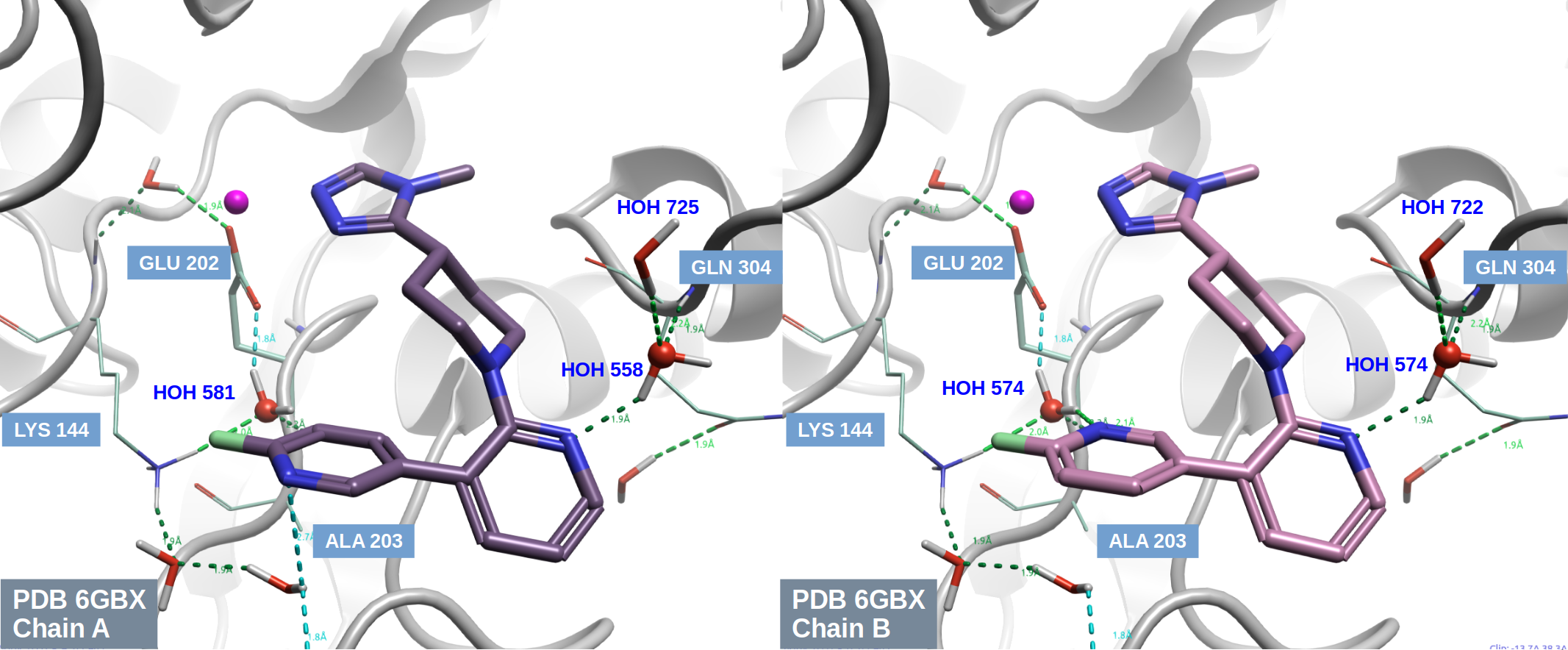
图3. 在PDB 6GBX中,配体SEN177的两个不同结合模式
在Chain B与C中,如图3右所示,HOH581(即HOH574)为水桥介导着蛋白与配体之间的氢键相互作用:一方面作为氢键供体与GLU202侧链羧基发生氢键作用,一方面作为氢键受体与ALA303主链酰胺NH发生氢键相互作用,同时作为氢键受体与LYS144末端的氨基阳离子发生氢键相互作用;另一方面作为氢键供体与SEN177的吡啶氮发生氢键相互作用。
而在Chain A中,HOH581的氧原子与配体2-氟吡啶环上碳原子之间的距离为3.1埃,这个距离使得HOH581受到配体排斥力量不是很强,有必要进一步对HOH581进行分析,并比较Chain A与Chain B的配体结合模式下该水的特性。
除了HOH581之外,还有一个靠近配体的结合水HOH558作为水桥介导了配体与蛋白的氢键网络。如图3左所示,HOH588一方面作为氢键供体与配体SEN177吡啶母核上的N原子发生氢键相互作用,另一方面作为氢键受体与蛋白GLN304骨架酰胺NH发生氢键相互作用,同时还与HOH725发生氢键相互作用。
与配体接触的水桥分子可以为基于结构的设计提供很多思路,分析它们的能量学特征有助于确定下一步的设计方案,因此采用GIST对结合口袋进行分析。
结合口袋的apo-GIST水分析
对Chain A的结合口袋进行apo-GIST水分析,水合位点由GIST水分子密度高的区域自然显现。结果如图4所示,包括HOH558与HOH581在内的大部分结晶水以及配体上与蛋白发生相互作用的氮原子都被高密度水(density\(\ge\)4)的等值图覆盖,这说明大部分实验结晶水被正确预测。
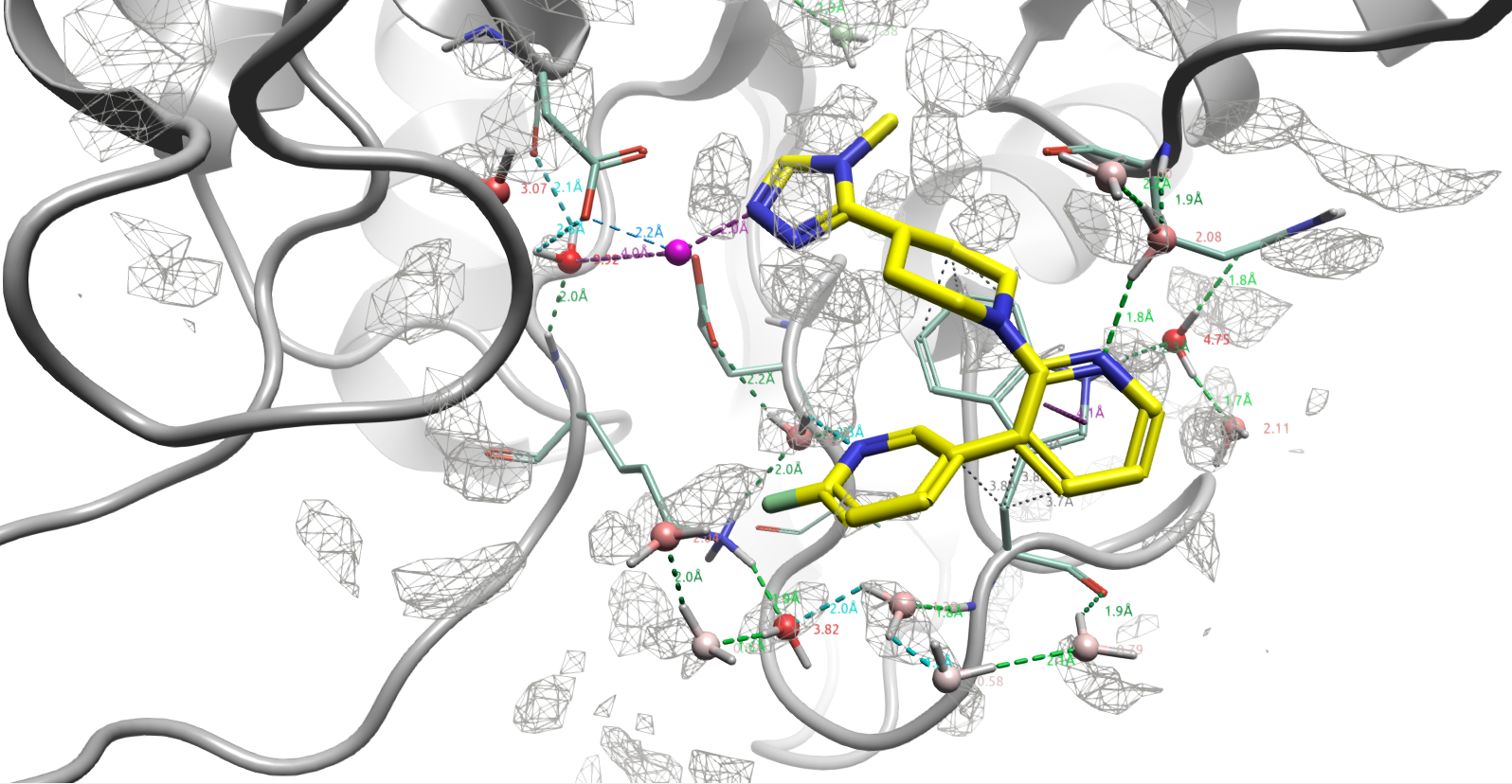
图4. 对PDB 6GBX的Chain A结合口袋进行apo-GIST分析的水密度图。网格:GIST Density\(\ge\)4 ;飘带:6GBX Chain A;黄色配体:SEN177;球状:距离SEN177 6 Å之内Chain A结晶水。
根据方程2(见方法部分),在不考虑方程3截断的情况下,用apo-GIST计算结果对距离SEN177 6 Å之内的Chain A实验结合水计算水合自由能,结果如图5所示。其中,HOH581的GIST ΔG = 2.31 kcal/mol,HOH558的GIST ΔG = 2.08 kcal/mol。
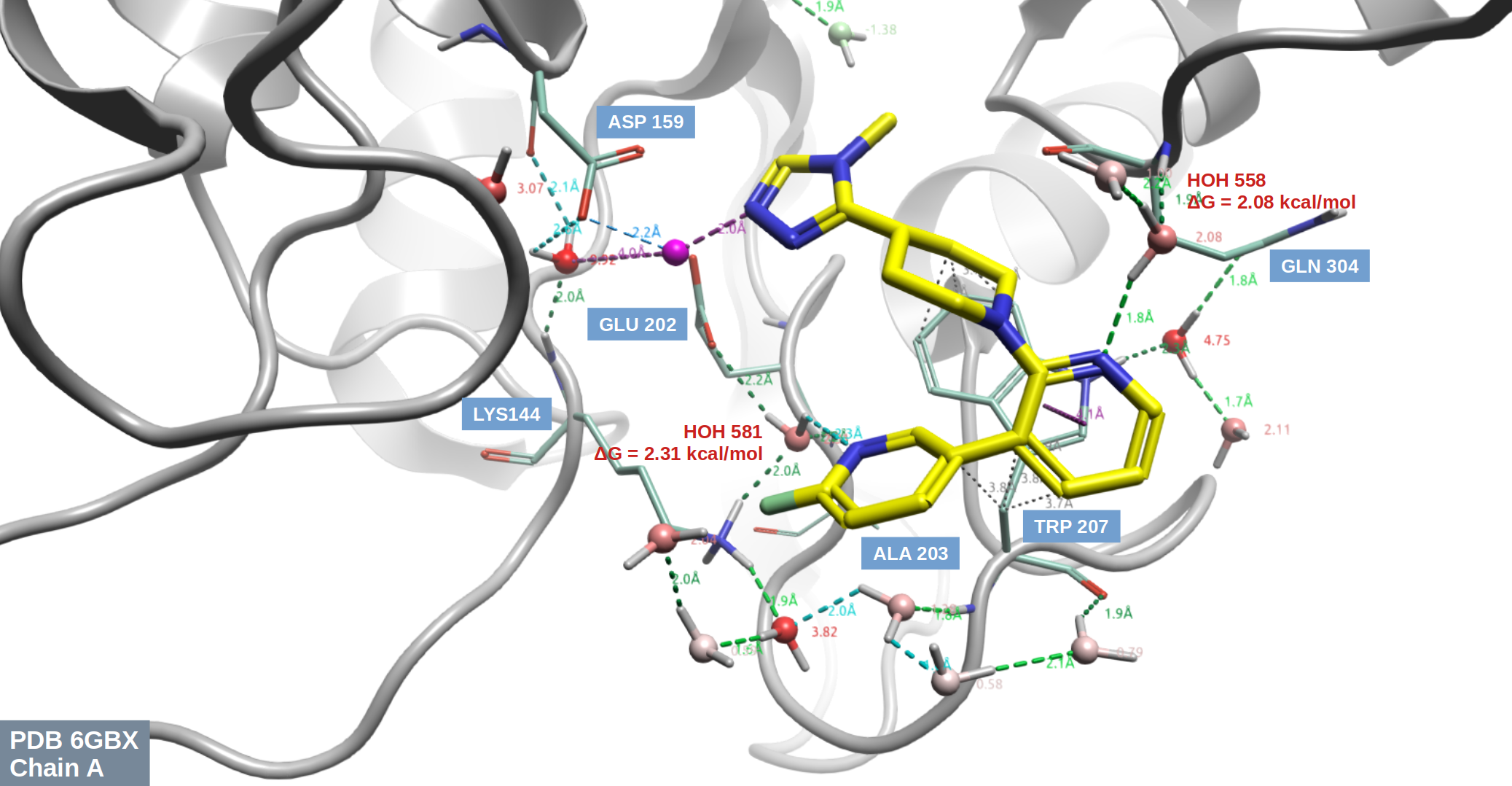
图5. Chain A结合口袋结合水的apo-GIST计算的水合自由能。飘带:6GBX Chain A;黄色配体:SEN177;球状:距离SEN177 6 Å之内Chain A结晶水,按apo-GIST计算的水合自由能着色。
对于桥连蛋白与配体的水有三种策略:替换、置换与保留。在Yu等人4等人的研究中,在SEN177母核吡啶引入腈基的设计是对HOH558这个桥连蛋白与配体氢键的Unhappy water进行了替换策略,让配体的腈基模拟水与蛋白直接相互作用。而对HOH581这个桥连蛋白氢键网络并且靠近配体的水,可以采用保留的设计策略,也就是将其视为蛋白的一部分,设计配体与这个水发生有利的相互作用。显而易见,Chain B的SEN177结合模式通过满足将HOH581作为蛋白一部分的保留策略,从这一点上看Chain B比Chain A的结合模式更具药物化学上合理性。Chain B的结合模式可解释表1的SAR:当R1保留间位氮吡啶基时,化合物4、5、9、28可以与HOH581发生氢键相互作用,具有两位数nM的IC50活性水平;当R1没有可与HOH581发生氢键相互作用的基团时,化合物6与7不能与HOH581发生氢键因而丧失了活性;形成鲜明对比的是,Chain A不能解释表1化合物的SAR。总的来说,对Chain A的apo-GIST分析可以初步认为Chain B是正确的结合模式,而Chain A的结合模式应该是错误的。
结合口袋的holo-GIST分析
对Chain A与Chain B两个结合模式分别进行holo-GIST分析(见方法部分),根据方程2(见方法部分)用holo-GIST计算的结果对SEN177周围6Å范围内的结晶水计算水合自由能,在不取方程3截断值的情况下,结果如图6所示。
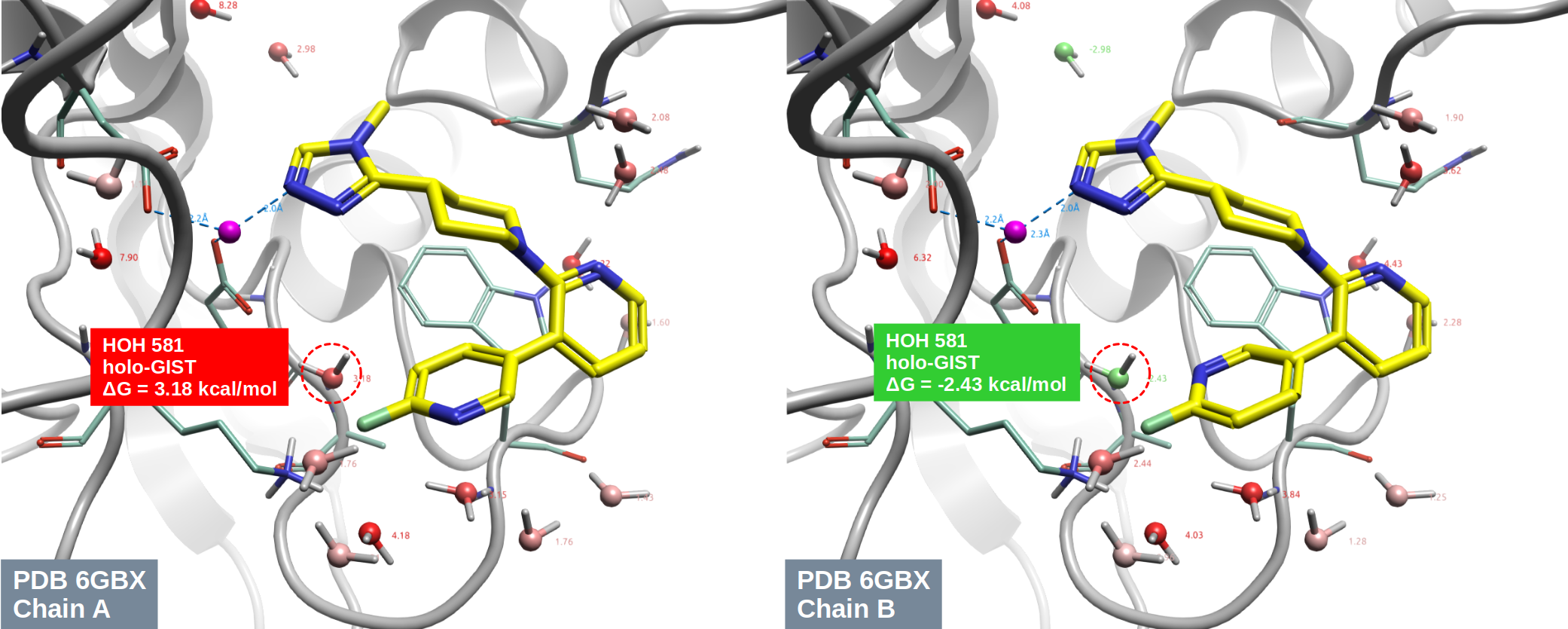
图6. PDB 6GBX共晶结构结合位点holo-GIST分析结果。左:Chain A结合模式;右:Chain B结合模式。球与细棍:结合水,按holo-GIST计算的水合自由能着色;红色圆圈高亮的水:HOH581。
图6红色圆圈高亮的HOH581,在Chain A结合模式(图6-左)下,holo-GIST水合自由能ΔG = 3.18 kcal/mol,这是一个高能的Unhappy水,意味着对蛋白与配体结合不利,起到去稳定化的作用。而在Chain B结合模式(图6-右)下,holo-GIST水合自由能ΔG = -2.43 kcal/mol,这是一个低能的Happy水,意味着对蛋白-配体的结合有利,起到稳定化的作用。总的来说,holo-GIST计算表明,从水合自由能角度看,Chain B结合模式再次能够合理的解释表1的SAR,而Chain A不能,因此Chain B结合模式是更加合理的。
MMGBSA计算
在Chain B结合模式下,用Flare MM/GBSA方法对表1化合物进行打分,结果表明,MM/GBSA-dG值与实验pIC50具有强线性相关性,Pearson R2=0.46,如图7所示。图7中圆圈大小表示pIC50的大小,可以发现非活性化合物6、7位于横坐标的dG正值区,活性化合物位于横坐标dG的负值区,活性与非活性化合物得到完美区分。
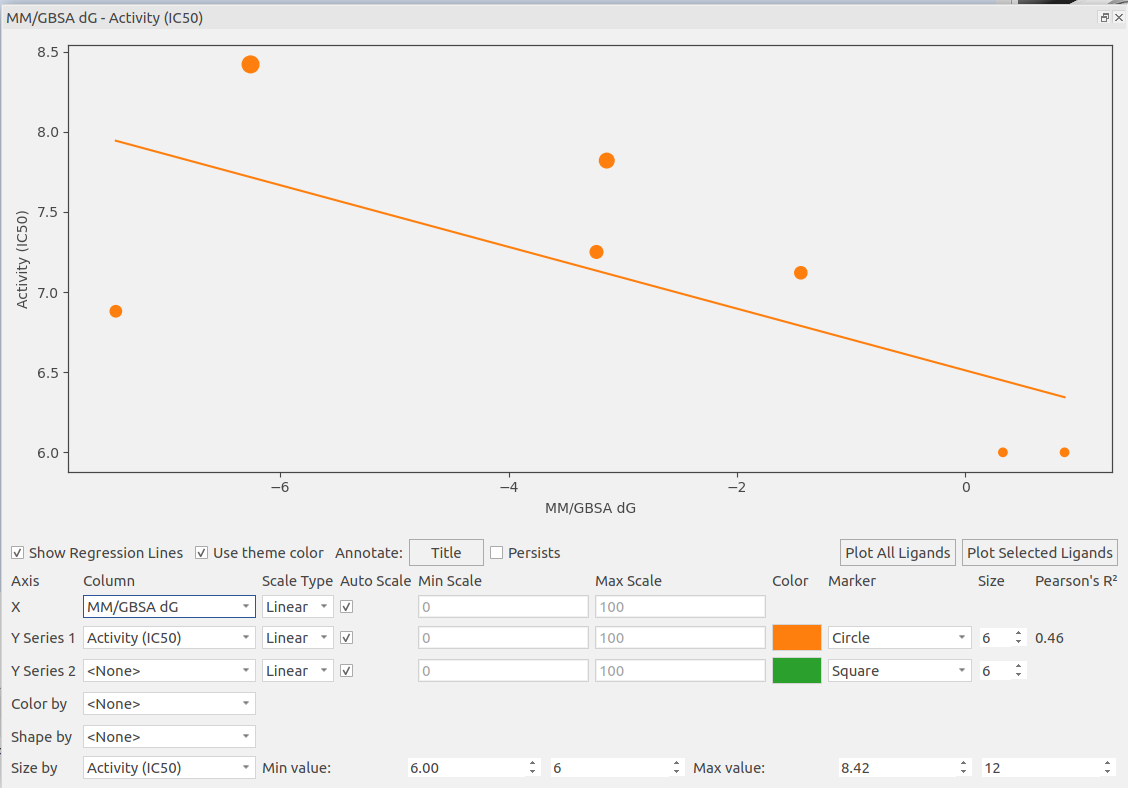
图7. 在Chain B结合模式下,MM/GBSA结合自由能预测值(MM/GBSA-dG)与实验pIC50具有强的相关性。
在Chain A结合模式下,如图8所示,此时MM/GBSA预测的结合自由能与实验pIC50不仅没有表现出任何的相关性,Pearson R2=0.003,而且也不能区分活性与非活性化合物。
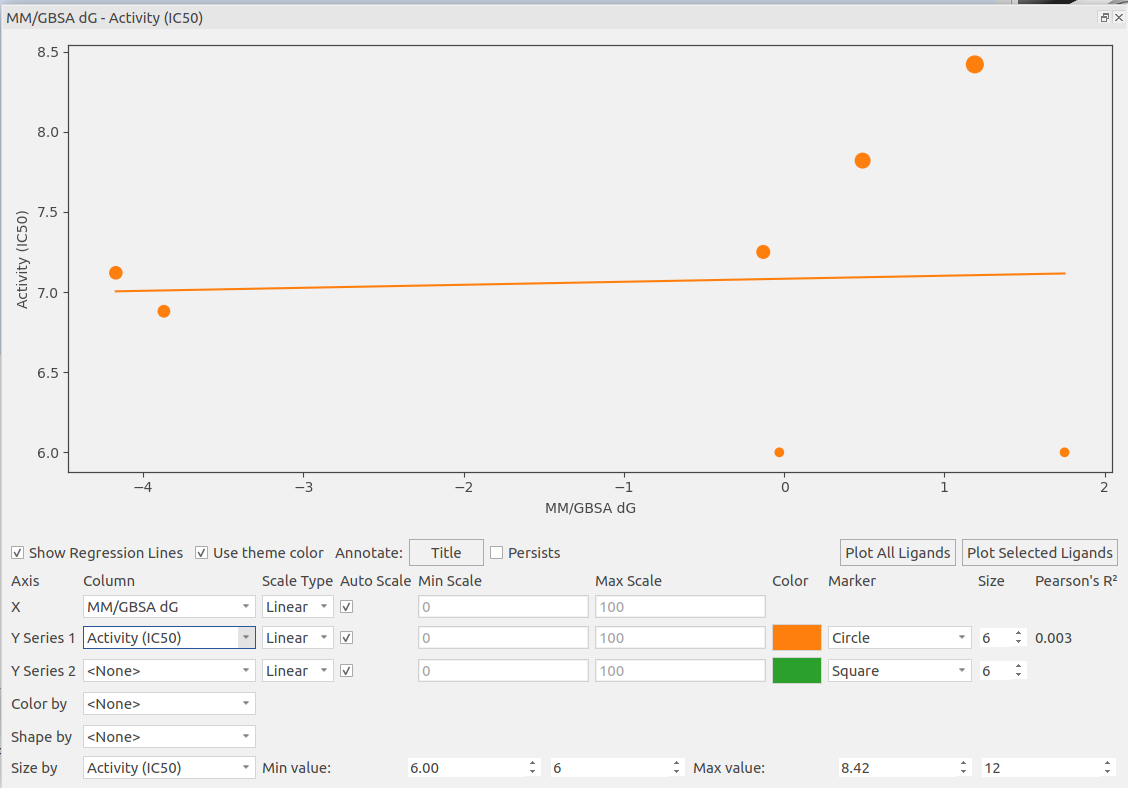
图8. 在Chain B结合模式下,MM/GBSA结合自由能预测值(MM/GBSA-dG)与实验pIC50没有表现出相关性。
需要注意的是,在Yu等人4的文章中并没有化合物6、7的IC50确切值。在进行结合自由能预测值与pIC50实验值相关性分析时,为了让分析可以进行,人为地将化合物6、7的IC50设置为1000nM。因此,上述的相关性分析并不是严格的定量分析,但是不妨碍定性的趋势与分类分析。
用QM分析HOH581与2-氟吡啶的相互作用
当然,上述的分析都是建立在HOH581与周围残基GLU202,ALA203,LYS144形成氢键网络的基础上,因为这个氢键网络会迫使HOH581的一个氢原子会大致指向SEN177的侧链吡啶,如图3所示。如果不考虑HOH581与蛋白的三个残基氢键网络,仅考虑HOH581与SEN177的侧链吡啶,则无论氮原子的取向如何HOH581与侧链吡啶均会发生“氢键”相互作用。这一点可以通过来量子化学计算来说明。为了方便计算,从准备好配体与蛋白结构(见材料与方法部分)中分别截取2-氟吡啶与HOH581作为相互作用研究用的模型分子(图9),对两个不同取向的体系用PSI4在bp86-d3/def2-tzvpp理论水平下固定C,N,O原子坐标进行真空状态下的结构优化,结果如图9所示。
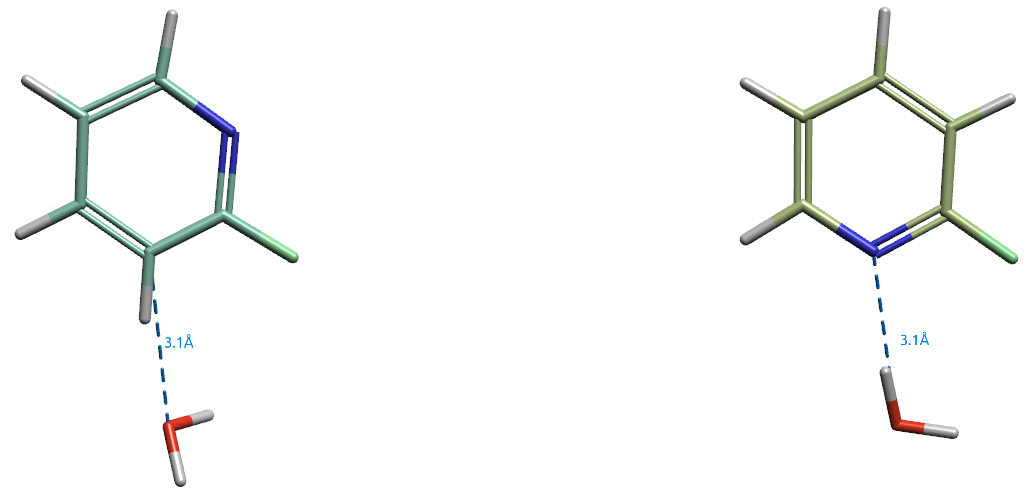
图9. 模型分子在BP86-D3/DEF2-TZVPP理论水平下QM结构优化的结果
我们可以看到,当2-氟吡啶为Chain A结合模式取向时(图9-左),吡啶环上的碳与HOH581的氧原子间距离为3.1埃,HOH581的两个氢远离吡啶,吡啶的C-H指向HOH581的氧原子,形成了C···H···O氢键。这个C···H···O氢键可以从isoValue=0.020 e/au3电子密度表面(图10-左)看出:在C-H与O之间形成了一个明显的电子密度桥。当2-氟吡啶为Chain B结合模式取向时(图9-右),吡啶C与HOH581的氧原子间距离为3.1埃,HOH581中的一个氢原子指向吡啶氮原子,这形成了经典的氢键相互作用。Chain B结合模式的isoValue=0.020 e/au3电子密度表面(图10-右)也可以清晰地看到吡啶氮原子与HOH581氧原子之间的电子密度桥。

图10. 模型分子的电子密度表面(isoValue=0.020 e/au3)。左:Chain A结合模式;右:Chain B结合模式
显然,在Chain A结合模式中,HOH581与2-氟吡啶发生C···H···O氢键相互作用(图9-左),HOH581的氢取向不利于与周围的蛋白残基形成氢键网络;而在Chain B结合模式中,HOH581与2-氟吡啶发生经典氢键相互作用(图9-右),HOH581的氢取向还可以与周围的蛋白残基形成氢键网络。用对称匹配微扰理论(SAPT)方法在sapt2+(3)dmp2/def2-tzvpp理论水平下计算上述优化过的2-氟吡啶与HOH581复合物的相互作用能(见材料与方法部分),结果表明HOH581与2-氟吡啶之间的C···H···O、HOH···N氢键取向的相互作用能分别为-1.385与-5.170kcal/mol。这说明经典氢键比C···H···O氢键的相互作用强的多,也就是说,Chain B结合模式的2-氟吡啶片段比Chain A结合模式的2-氟吡啶片段在与HOH581发生相互时,具有更有利的相互作用能。
综合结合位点apo/holo-GIST水分析结果、MMGBSA结合自由能计算、QM计算以及现有化合物的SAR,可以推定:在Chain A中SEN177的结合模式应该是错误的,而在Chain B、C链的SEN177结合模式是正确的。选择正确的结合模式对后续的分子设计至关重。
SPARK水分子替换:重现QP5020的设计
根据前面的apo-GIST计算,HOH558是“unhappy”水,其apo-GIST ΔG = 1.85 kcal/mol,因此具备可替换的热力学基础;同时也可以起到“水桥”的作用,介导了配体与受体的氢键网络。替换此类水并与蛋白形成氢键相互作用可以增强配体的结合亲和力。替换实验用Spark来实现,图11展示了水分子替换实验的规划:高亮部分显示了需要被替换的分子片段与水。

图11. 用Spark替换吡啶母核与Unhappy water-HOH558的实验规划。
将共晶配体与HOH558作为替换实验的起始分子,如图12所示,高亮部分为要被替换的部分,链接点1、2的原子设置为芳香环原子以便使得结果接近起始分子。
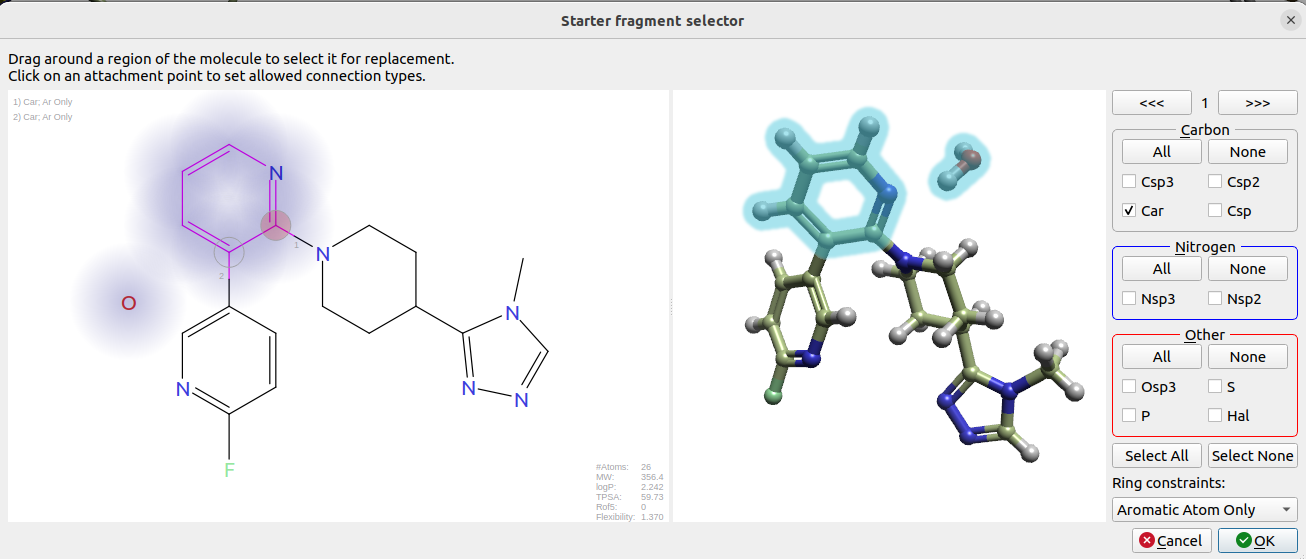
图12. 替换实验的起始分子
为了更好的模拟水分子,如图13所示,设置了两个约束条件:1)在HOH558氧原子处添加氢键受体的药效团约束;2)在HOH558氧原子的负静电场点处添加场约束。
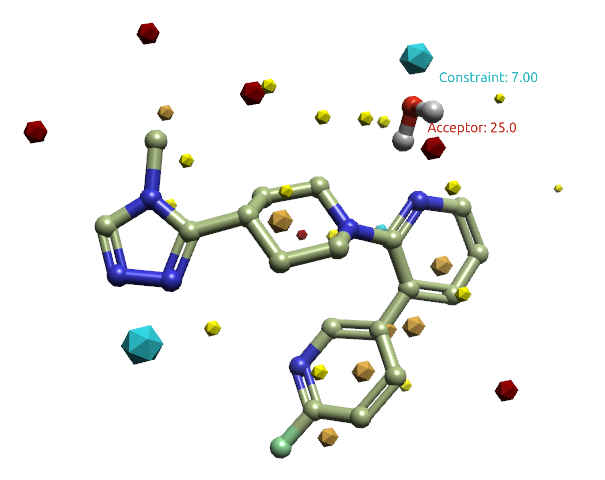
图13. 替换实验的约束设置
在这个替换实验里,对ChEMBL_common,VeryCommon与Common三个数据库共161854个分子片段进行了虚拟筛选,如图14所示。
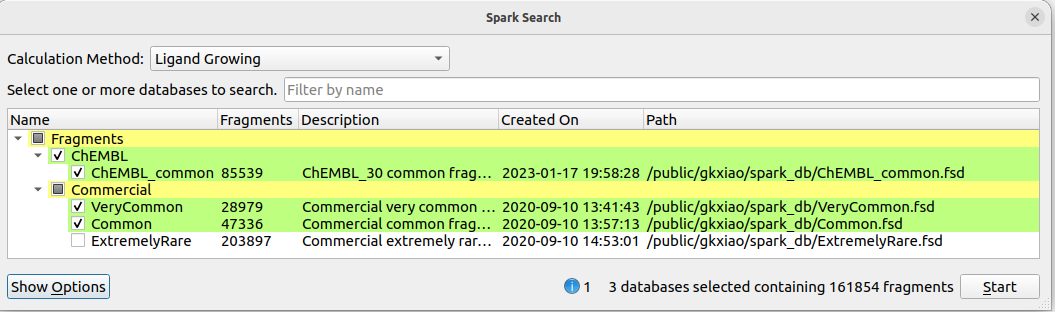
图14. 替换实验的数据库设置
打分函数采用了基于配体相似性的方法,其中形状与场相似性比例为50:50,如图15所示,最后保留打分值最高的500个结果。

图15. 替换实验的打分函数
计算完毕,命中500个结构,共73种不同的化学类型骨架。打分最高的骨架类别为苯环,总共命中苯环衍生物361个。其中包含了Yu等人4报道化合物4(QP5020),其在500个结果里排名第2。QP5020与起始分子的Shape score = 0.917,Field score= 0.862。场点比较如图16所示,可以看到苯甲腈的CN方向上有一个蓝色的场点与起始分子水的蓝色场点一致。
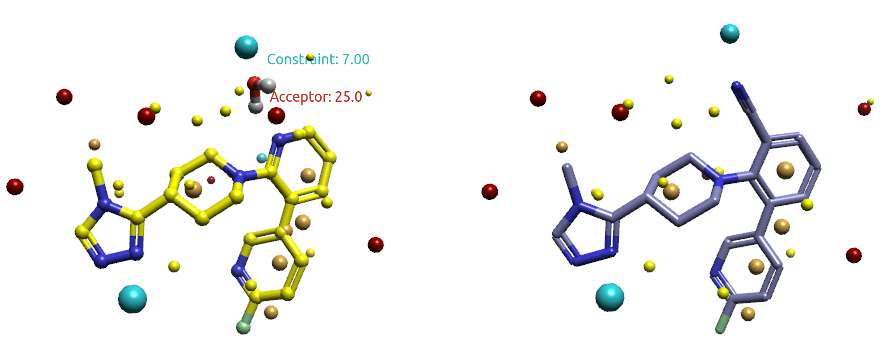
图16. 起始分子(左)与替换实验排名第2结果(QP5020,右)的场点比较
用Flare Dock在‘Very Accurate but Slow’模式下将替换实验排名第2(QP5020)的结果对接到6GBX的结合位点,对接时将Zn与“Happy water”-HOH581作为蛋白的一部分。对接打分最佳的结合模式重现了Spark生成的Pose,如图17所示,苯甲腈片段替换吡啶母核之后,腈基替换了HOH558并与GLU304的主链酰胺NH发生氢键相互作用,其中侧链吡啶氮与“Happy water”-HOH581发生氢键相互作用。这个相互作用是与Yu等人4描述的结合模式最大区别所在。
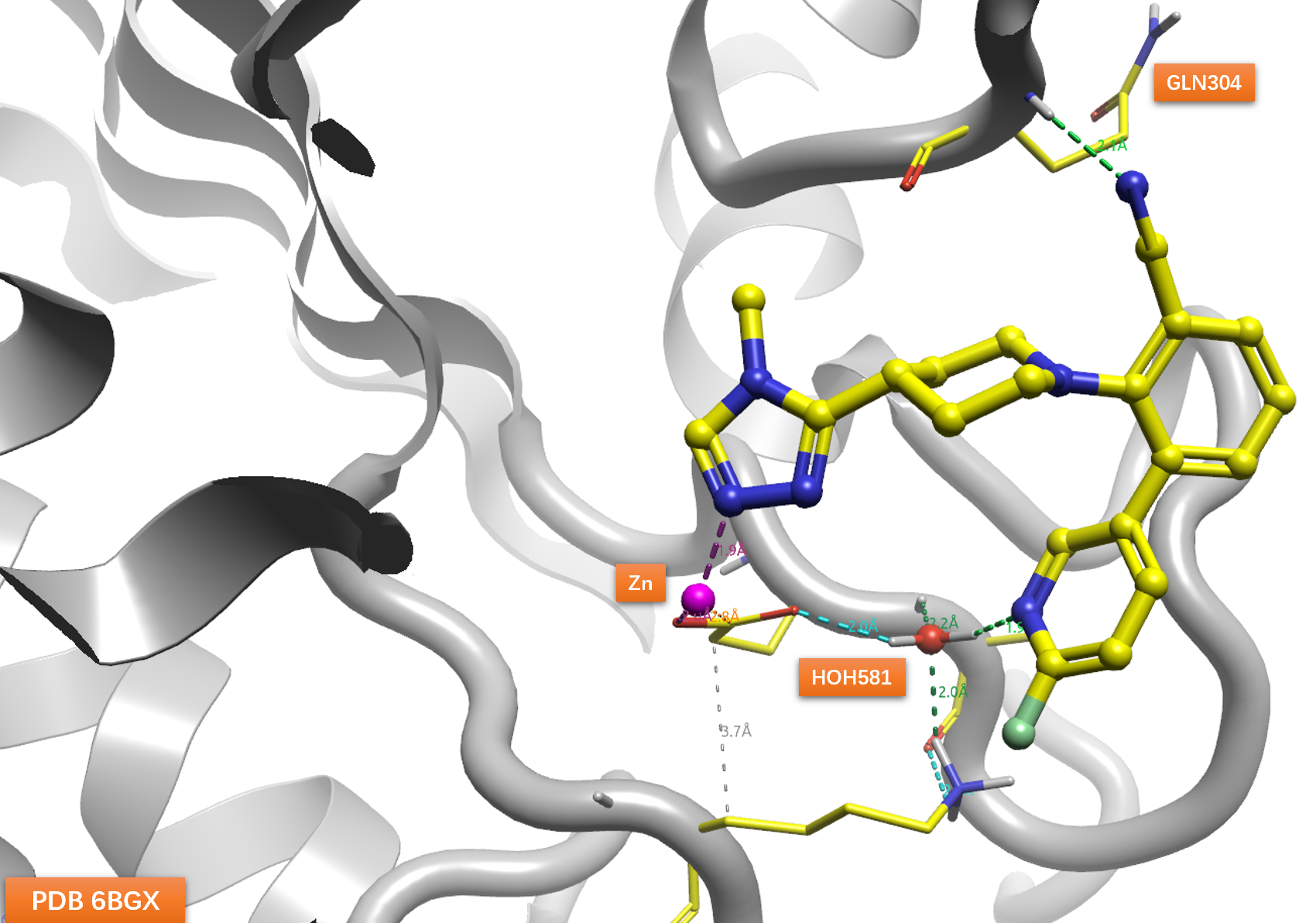
图17. 替换实验排名第2结果(QP5020)的redocking结合模式
总的来说,Spark的水分子替换实验以高分(排名第2)的方式将苯甲腈片段识别为吡啶母核的替换结构,命中了Yu等人4报道的化合物4(QP5020)。该化合物与起始化合物(包括水在内)具有高度相似的形状与静电场,对接实验可以重现预期的相互作用模式。在与徐锡明博士交流的时候,一致认为这是个相当完美的设计,并入选到下一轮的优先性排序里5。
XED场点分析发现Zn结合片段的优化机会
可视化分析化合物4的XED场点,如图18-左所示,可以发现N-甲基三氮唑的蓝色负场点偏离Zn离子,这提示了化合物4的三氮唑与Zn的结合模式存在不利的几何而可能对活性不利,因此这里有潜在的优化机会,有必要用QM进一步确认。相比之下,化合物28(QP5038)的N-甲基咪唑的蓝色负场点与Zn离子重合,如图18-右所示,N-甲基咪唑与Zn具有更有利的静电相互作用。
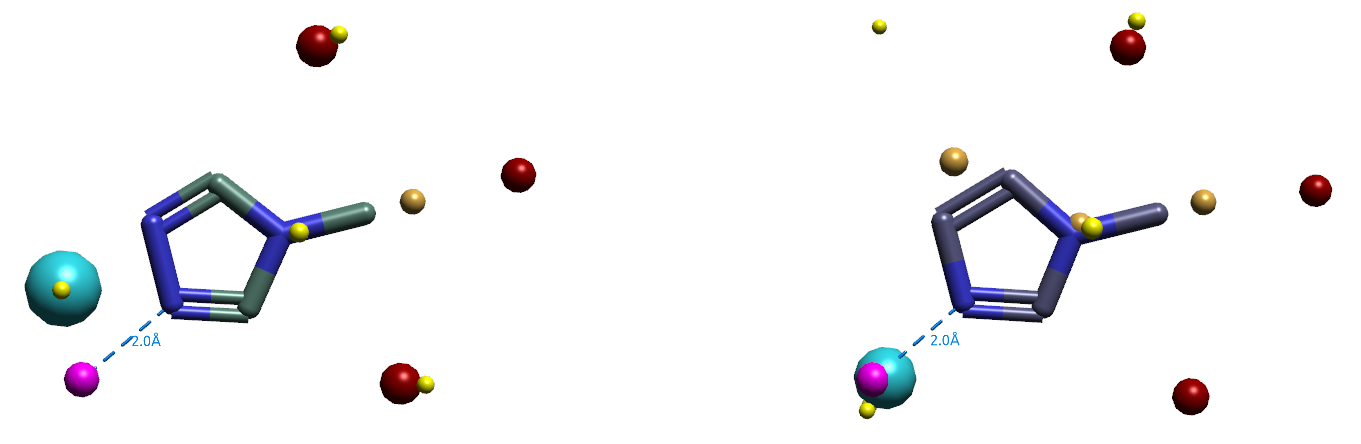
图18. Zn结合片段的XED场点。左:N-甲基三氮唑(代理QP5020,化合物4); 右:N-甲基咪唑(代理QP5038,化合物28); 粉红色球:Zn2+离子
用QM方法在APFD/6-311+G(d,p)理论水平对Zn结合片段进行了几何优化(详见材料与方法部分),结果如图19所示:N-甲基三氮唑-Zn的几何发生显著的变化,角Zn-N3-N4由结合模式的~116°变为69°;相比之下N-甲基咪唑-Zn的几何没有发生显著的变化,角Zn-N3-C4由由结合模式的116°变为125°。这说明,与Zn离子结合时,QP5020(N-甲基三氮唑片段)要比QP5038(N-甲基咪唑片段)消耗更多的结合能去维护结合模式,导致N-甲基三氮唑比N-甲基咪唑片段不利于与QPCTL结合。
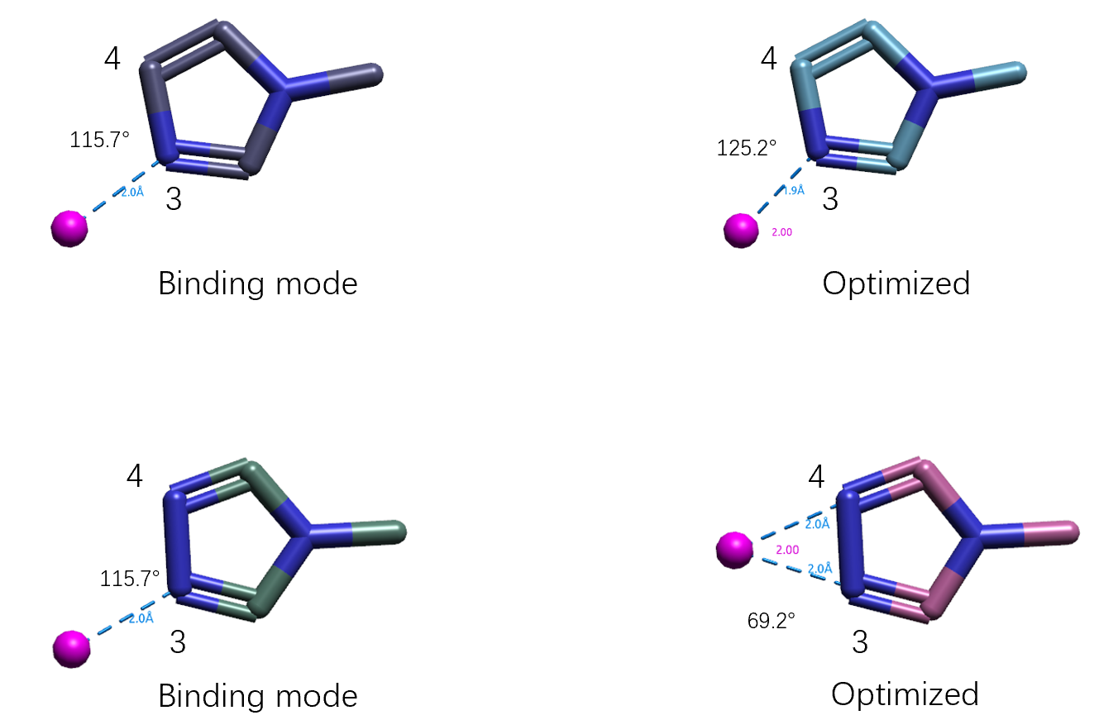
图19. Zn结合片段的QM几何优化。上:N-甲基咪唑(代理QP5038);下:N-甲基三氮唑(代理QP5020)。左:结合模式的几何;右:QM优化的几何
QM几何优化的结果与XED场点分析是相一致的:N-甲基三氮唑-Zn的QM几何优化结果表明,Zn出现在了XED蓝色负场点处。这从计算上证实了Zn离子与三氮唑并没有达到最好的相互作用几何。而N-甲基咪唑-Zn在基于QM的几何优化之后Zn离子位置几乎没有变化,这意味着Zn离子与N-甲基咪唑有着较好的相互作用几何。因为XED场点的计算比起QM来说要快的多,所以它的另一个重要用途是快速发现静电不匹配的片段,以发现优化的机会,比如这里的发现N-甲基三氮唑Zn离子结合片段的优化机会。
用QM评估N-甲基三氮唑、N-甲基咪唑以及咪唑等结合片段与Zn离子的相互作用能
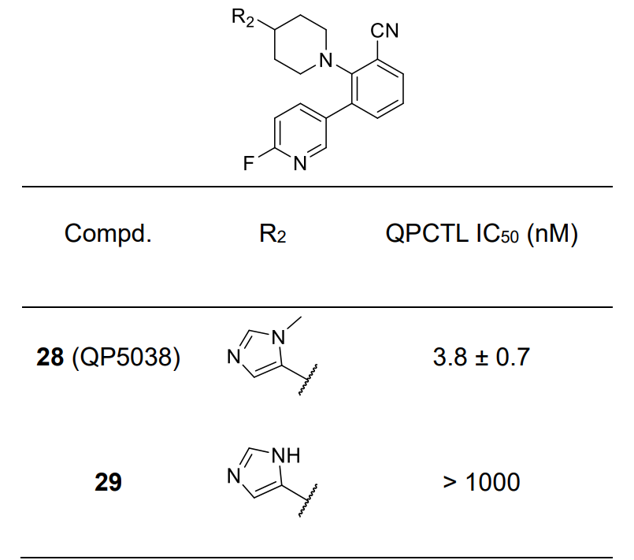
在Yu等人4的研究中,化合物28(QP5038)与29的唯一区别在于Zn离子结合片段:前者为N-甲基咪唑,后者为咪唑,见表2。28、29化合物的一个甲基差异引起了260多倍的IC50差异,这是一个标准的活性悬崖现象,可归因于两个片段与Zn的相互作用能差异。接下来我们用QM方法来计算不同结合片段与Zn离子的相互作用能,并用计算结果来解释28与29的活性悬崖。
表2. 化合物28与29的QPCTL体外活性4
基于QM的相互作用分析可以定量计算分子间的相互作用能。在BP86-D3/DEF2-TZVP理论水平下对N-甲基三氮唑、N-甲基咪唑、咪唑进行几何优化,然后用Flare叠合到PDB 6GBX的SEN177的N-甲基三氮唑获得它们的结合模式。然后使用PSI49,10实现的对称匹配微扰理论(SAPT)方法在sapt2+(3)dmp2/def2-tzvpp水平下计算模型分子N-甲基三氮唑、N-甲基咪唑、咪唑与Zn之间的相互作用能(见材料与方法部分),结果如表3所示。
表3. N-甲基三氮唑、N-甲基咪唑、咪唑与Zn2+离子的SAPT相互作用能分析结果
| Items | N-甲基三氮唑 | N-甲基咪唑 | 咪唑 |
|---|---|---|---|
| Electrostatics | -113.850 | -121.096 | -114.4423 |
| Exchange | 48.250 | 52.648 | 56.863 |
| Induction | -92.950 | -101.069 | -95.706 |
| Dispersion | -5.088 | -5.340 | -5.653 |
| Total SAPT2+(3)dMP2 | -163.638 | -174.857 | -158.940 |
结合片段-Zn相互作用能对抑制剂的活性影响可以被实验证实,N-甲基咪唑比脱去甲基之后的咪唑与Zn的结合能增加了19.5 kcal/mol,由-174.857 kcal/mol变化为-158.940 kcal/mol(见表3),相应地化合物活性也降低,对QPCTL的体外抑制活性IC50由28的3.8 nM变化为在1000 nM水平测不到29的抑制活性,见表2。
QM方法可以用来对不同的Zn结合片段进行优先级排序。比之N-甲基三氮唑与咪唑,N-甲基咪唑与Zn具有更有利的静电相互作用(Electrostatics)与诱导相互作用(Indution),这两项有利的相互作用远大于不利的排斥项(Exchange),最终导致N-甲基咪唑比N-甲基三氮唑、咪唑具有更强的Zn离子相互作用能。结合片段-Zn相互作用能与抑制剂的活性有直接的关系,相互作用能越强,抑制活性越强。
Zn结合片段N-甲基三氮唑的生物等排体替换:重现QP5038的设计
上一节的QM计算与XED场分析表明,PDB 6GBX的N-甲基三氮唑与Zn的结合模式并非低能几何构象,因此与Zn结合更好的片段有望提高结合亲和力,其要点是新片段的负场点与Zn重合,这可以用来指导后续的Spark生物等排体替换实验。
N-甲基三氮唑的替换用Spark V10.7的等排体替换工作流6来实现,起始分子同前面QP5020的设计小节,并且将蛋白结构作为体积约束使用,如图20所示,连接点1的原子类型设置为:Csp3、Csp2、Car、Nsp3、Nsp2 and ring atom only。
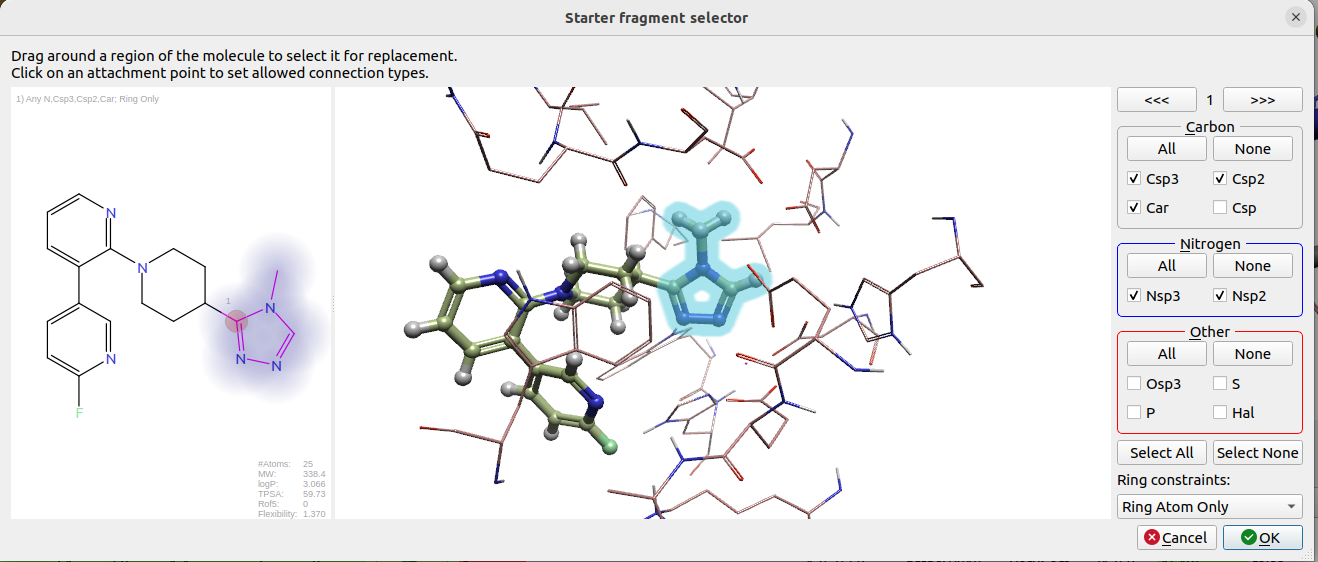
图20. Zn离子结合片段的生物等排体替换起始分子及参数设置
为了让新化合物能与Zn离子具有更好的相互作用模式,在约束编辑器里使用“Move”工具将N-甲基三氮唑的蓝色负场点移动至Zn离子上,如图21所示。
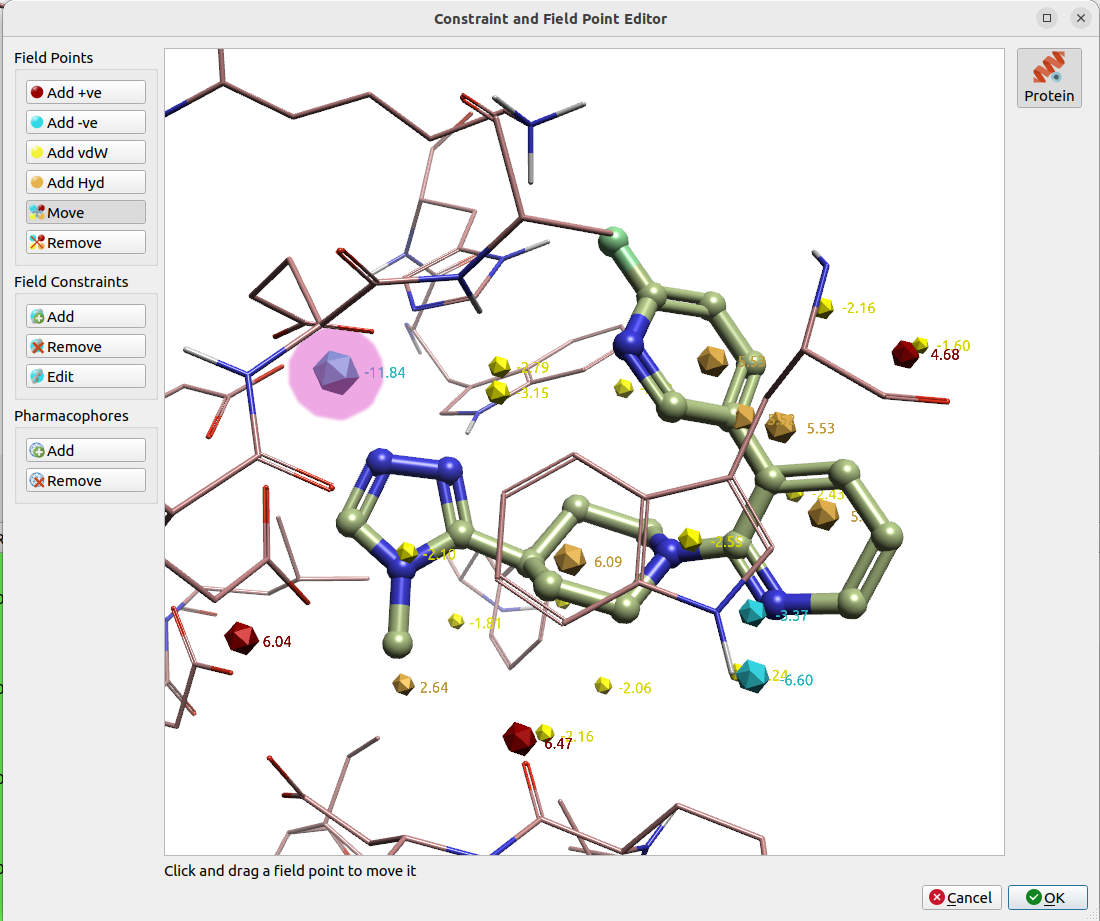
图21. Zn离子结合片段的生物等排体替换的约束设置
然后对ChEMBL_common,VeryCommon与Common三个数据库共161854个分子片段进行了虚拟筛选,如图14所示,其它参数同QP5020的设计小节。结果命中了500个结构,共包含152种不同化学骨架类型。按骨架打分从高到低排序,打分最高的化学骨架是三氮唑本身、排序第2的为四氮唑、排序第3的为咪唑,如图22所示。

图22. Zn离子结合片段的生物等排体替换实验结果。左上:起始分子SEN177;左下:Rank#1;右上:Rank#3;右下:Rank#2
在打分最高的化学类型中,只有排名第2的咪唑类片段的蓝色负静电场点满足预设的要求,以此为过滤条件,N-甲基咪唑是替换实验首选的结果,这正是Yu等人4报道QP5038的Zn结合片段。
总的来说,根据N-甲基三氮唑-Zn的QM几何优化与结合模式的XED场分析结果来指导Spark生物等排体替换实验可以在打分最靠前位置命中N-甲基咪唑作为新的Zn结合片段,并被Yu等人4报道的QP5038所验证。Spark还命中其它尚未被文献验证的高分Zn结合片段,这些片段有望发展出新的系列化合物。
材料与方法
蛋白结构的准备
鉴于QPCTL与QPCT的结合口袋具有保守性,因此直接以QPCT-SEN177的共晶结构PDB 6GBX作为QPCTL-SEN177结合模型的代理进行分析。将共晶结构PDB 6GBX下载到Flare7中,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,任何截短的蛋白质链被封端作为蛋白质准备的一部分。
注意到PDB 6GBX包含A、B、C三个链,A链配体的构象与B、C链的不同,如图23所示:A链配体的侧链2-氟吡啶氮不与HOH581形成氢键相互作用;B、C链配体的侧链2-氟吡啶发生翻转,使得吡啶氮与水发生氢键相互作用。将Chain A、B、C拆成3个不同的蛋白,将Chain B、C叠合到Chain A上,以使得后续的GIST结果在可视化时具有可比性。

图23. PDB 6GBX进行结构准备之后,A链配体与B、C链配体的结合构象是不同的。左:A链;中:B链;右:C链
由于配体具有两种可能的结合模式,因此有必要论证哪个结合模式是正确的。在本研究中,我们暂且先假设链A的配体构象是正确的,在后续的研究中将会证明这个假设是错误的,而B、C链中地配体结合模式才是对的。将配体SEN177从蛋白中提取出来到配体表单,删除Other里面的硫酸根离子与乙二醇(EDO)而仅保留Zn离子。
配体结构的准备
以上一步蛋白结构准备提取出的配体SEN177为模板,用Flare7在结合口袋里编辑SEN177,准备出表1的其它化合物结构,并在蛋白背景下用力场进行能量最小化。
GIST分析
以Chain A为例,对结合位点进行apo-GIST分析,使用了如下条件:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Protein: 6GBX_A
- Chains: A Chain, A Other,A Water
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
- GCNCMC: During equlibration only with buffer 4.00 Å
在进行apo-GIST分析时,Ligand选None;在进行holo-GIST分析时,Ligand选应的配体。
水合位点预测
对水密度进行聚类分析得到显式的水合位点。首先,选择具有最高水氧密度的体素来确定第一个水合位点的位置。然后排除所有在第一个水合位点2.5Å范围内的体素,不再考虑。然后,对下一个最高密度的体素重复此过程,直到没有剩下密度高于2倍本体水氧密度的体素为止。计算采用基于pyflare的脚本gist_hydrate_sites.py来实现。
水合自由能计算
然后识别每个水合位点周围1.4Å半径内的体素(Voxel):
$$
V = \left\{v_{i} | v_{i} \in hydration\ site\right\} \cdots(1)
$$
如方程1所示,如果一个体素包含在水合位点的范德华半径内,则累加这些体素的能量,并将总和乘以体素的体积(vol = 0.125 Å3),得到以kcal/mol为单位的值,如方程2所示:
$$
ΔG = vol \times \sum_{v_{i} \in V}G_{GIST}(v_{i})\cdots(2)
$$
方程2的ΔG即是水合位点的水合自由能。在计算的时候,可选地对体素vi上的GGIST值设置了上限截断值+3kcal・mol-1・Å-3以去掉因为GIST算法带来的极端高值的体素,如方程3所示:
$$
G_{GIST}(v_{i}) = \left \{
\begin{aligned}
& +3\ &if\ G_{GIST}(v_{i}) \ge +3 \\
& G_{GIST}(v_{i}) &\ otherwise
\end{aligned}
\right.\cdots(3)
$$
方程2的水合自由能采用pyflare脚本(gist_watscore.py)实现,并将水合自由能记录在PDB的Temperature列。
用Spark进行水分子替换
用Spark V10.76来进行基团替换实验,这是一个以被充分验证的方法8。在本文中,我们采用Spark的等排体替换工作流来进行水分子替换,其中起始分子与要替换的水分子是在从前面的蛋白结构准备与配体结构步骤得到。
起始分子:starter.sdf
MM/GBSA预测结合自由能
用Flare MM/GBSA7来对表1化合物进行单点结合自由能预测。在预测SEN177时将HOH581,HOH558与Zn作为蛋白的一部分;而在预测4,5,6,7,9与28时将HOH581与Zn作为蛋白的一部分。其它参数如下:
- Calculation method: Normal
- Solvent Model: Implicit | Implicit GBn2
- Minimize: Protein and Ligand
在Yu等人4的文章中并没有化合物6、7的IC50确切值,在进行IC50的预测值与实验值相关性分析时,为了让分析可以进行,人为地将化合物6、7的IC50设置为1000nM。
2-氟吡啶与HOH581复合物的电子密度表面计算
用Flare QM在B3LYP-D3BJ/6-311G(d,p)理论水平对2-氟吡啶与HOH581的复合物进行单点计算,然后在Flare Surfaces工具里调整QM electron density的isoValue值为0.020获得图10的电子密度表面。
对称匹配微扰理论(SAPT)计算相互作用能
使用PSI49,10实现的对称匹配微扰理论(SAPT)方法在sapt2+(3)dmp2/def2-tzvpp水平下计算模型分子2-氟吡啶与HOH581之间的相互作用能。
2-氟吡啶(PDB 6GBX构象,图10-左)与HOH581的相互作用能计算:
memory 24 GB molecule dimer { 0 1 C -2.00370000 -1.16990000 -0.02770000 C -0.61860000 -1.46650000 0.11490000 C 0.35050000 -0.49820000 0.18040000 C -0.02670000 0.79110000 0.07490000 F 0.85570000 1.79640000 0.11420000 N -1.34490000 1.15130000 -0.08210000 C -2.35900000 0.16330000 -0.15240000 H -0.31910000 -2.50640000 0.18990000 H -3.36380000 0.54010000 -0.28730000 H 1.39380000 -0.76300000 0.28840000 H -2.73750000 -1.96520000 -0.07190000 -- 0 1 O 3.37820000 -0.71960000 -0.15730000 H 4.16130000 -0.66360000 0.38690000 H 3.49830000 -0.03060000 -0.84180000 } set { scf_type DF freeze_core True basis def2-tzvpp } energy('sapt2+(3)dmp2') |
用24核心执行计算:
psi4 dimer.com dimer.log -n 24 |
结果:
Total SAPT2+(3)dMP2 -2.20648055 [mEh] -1.38458745 [kcal/mol] -5.79311388 [kJ/mol] |
2-氟吡啶(翻转构象,图10-右)与HOH581的相互作用能计算:
memory 24 GB molecule dimer{ 0 1 C 1.98260000 -1.15820000 0.01660000 C 0.59980000 -1.46960000 -0.11710000 C -0.01680000 0.78110000 -0.06230000 F -0.99700000 1.68630000 -0.11530000 C 2.32400000 0.17830000 0.14480000 H 0.25210000 -2.49210000 -0.20130000 H 3.35210000 0.49540000 0.26780000 H 2.71760000 -1.95190000 0.04460000 C 1.29850000 1.15530000 0.08660000 H 1.54700000 2.20650000 0.17480000 N -0.38040000 -0.51190000 -0.17080000 -- 0 1 O -3.40280000 -0.76860000 0.18830000 H -3.69280000 0.12960000 0.00530000 H -2.44560000 -0.75330000 0.02360000 } set { scf_type DF freeze_core True basis def2-tzvpp } energy('sapt2+(3)dmp2') |
用24核心执行计算:
psi4 dimer-altaN.com dimer-altaN.log -n 24 |
结果:
Total SAPT2+(3)dMP2 -8.23952745 [mEh] -5.17038154 [kcal/mol] -21.63287635 [kJ/mol] |
Zn-结合片段的QM几何优化
N-甲基三氮唑-Zn的3D结构(图24-左)是用Flare从PDB 6GBX的结合位点里截取出来,然后对副本进行修改得到N-甲基咪唑-Zn的3D结构(图24-右)。
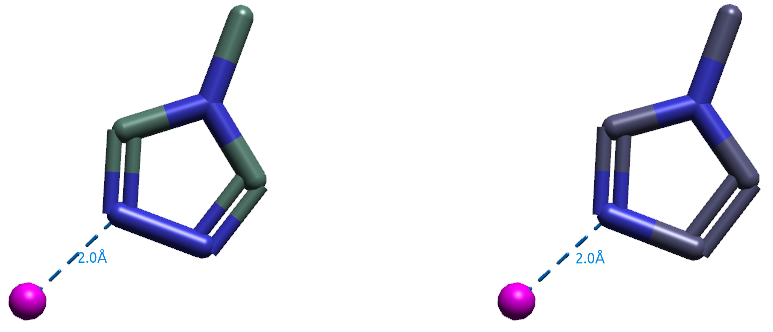
图24. N-甲基三氮唑-Zn(左)与N-甲基咪唑-Zn(右)的3D结构
几何优化使用Gaussian 16 A.01在APFD/6-311+G(d,p)理论水平下进行,其中N-甲基三氮唑-Zn的几何优化输入文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | %chk=triazole_opt.chk # opt apfd/6-311+g(d,p) Structure: 6GBX-Ligand Triazole-Zn complex; Job: Opt at APFD/6-311+G(2d,p) level 2 1 C 24.49500000 5.16500000 34.26200000 N 23.02600000 4.82000000 34.44200000 C 22.36500000 3.80600000 33.86200000 N 21.11200000 3.88000000 34.24600000 N 20.93900000 4.92800000 35.07000000 C 22.13800000 5.52500000 35.15600000 H 24.96100000 4.41800000 33.78800000 H 24.58200000 5.99900000 33.71800000 H 24.90900000 5.30500000 35.16100000 H 22.75600000 3.11900000 33.24900000 H 22.42790000 6.35120000 35.63910000 Zn(PDBName=ZN,ResName=ZN,ResNum=401_A) 19.56200000 2.58500000 34.19800000 !a blank line |
其中N-甲基咪唑-Zn的几何优化输入文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | %chk=imidazole_opt.chk # opt apfd/6-311+g(d,p) Structure: 6GBX-Ligand Imidazole-Zn complex Job: Opt at APFD/6-311+G(d,p) level 2 1 C 24.49500000 5.16500000 34.26200000 N 23.02600000 4.82000000 34.44200000 C 22.36500000 3.80600000 33.86200000 N 21.11200000 3.88000000 34.24600000 C 22.13800000 5.52500000 35.15600000 H 24.96100000 4.41800000 33.78800000 H 24.58200000 5.99900000 33.71800000 H 24.90900000 5.30500000 35.16100000 H 22.75600000 3.11900000 33.24900000 H 22.42790000 6.35120000 35.63910000 Zn(PDBName=ZN,ResName=ZN,ResNum=401_A) 19.56200000 2.58500000 34.19800000 C 20.93900000 4.92800000 35.07000000 H 20.06349907 5.27751199 35.57620103 !a blank line |
N-甲基三氮唑、N-甲基咪唑、咪唑等结合片段与Zn的相互作用能分析
在BP86-D3/DEF2-TZVP理论水平下对N-甲基三氮唑、N-甲基咪唑、与咪唑进行几何优化,然后在Flare里叠合到PDB 6GBX共晶配体SEN177的N-甲基三氮唑上,然后使用PSI49,10实现的对称匹配微扰理论(SAPT)方法在sapt2+(3)dmp2/def2-tzvpp水平下计算模型分子与Zn之间的相互作用能。
N-甲基三氮唑-Zn(图18-左)的相互作用能计算:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 | memory 24 GB molecule dimer { 0 1 C 24.45970 5.14230 34.28400 N 23.05170 4.85230 34.41360 C 22.35590 3.82100 33.86740 N 21.10810 3.85120 34.24470 N 20.96170 4.93210 35.07230 C 22.12720 5.51020 35.16090 H 24.80910 4.81980 33.30360 H 24.61970 6.21680 34.36970 H 25.03920 4.63170 35.05590 H 22.79990 3.09130 33.20730 H 22.35250 6.39490 35.73710 -- 2 1 Zn 19.562 2.585 34.198 } set { scf_type DF freeze_core True basis def2-tzvpp } energy('sapt2+(3)dmp2') |
结果:
Total SAPT2+(3)dMP2 -163.63803595 [kcal/mol] |
N-甲基咪唑-Zn(图18-右)的相互作用能计算:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | memory 24 GB molecule dimer { 0 1 C 24.46620 5.14310 34.26600 N 23.03840 4.83050 34.42980 C 22.39560 3.82700 33.86210 N 21.09780 3.87040 34.25410 C 22.15850 5.55110 35.20930 C 20.94250 4.96280 35.10930 H 24.91810 4.41230 33.59780 H 24.55860 6.14210 33.83930 H 24.94780 5.09920 35.24310 H 22.84540 3.10290 33.19890 H 22.46690 6.42150 35.77070 H 20.00530 5.23460 35.57120 -- 2 1 Zn 19.562 2.585 34.198 } set { scf_type DF freeze_core True basis def2-tzvpp } energy('sapt2+(3)dmp2') |
结果:
Total SAPT2+(3)dMP2 -174.85693784 [kcal/mol] |
咪唑-Zn的相互作用能计算:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 | memory 24 GB molecule dimer { 0 1 N 23.03830 4.82900 34.44550 C 22.34030 3.81130 33.87250 N 21.07890 3.83430 34.22110 C 20.95140 4.91360 35.05600 C 22.15390 5.54860 35.21140 H 22.80290 3.08950 33.21520 H 22.45410 6.41580 35.77690 H 20.00160 5.17370 35.49910 H 24.01930 5.02280 34.32740 -- 2 1 Zn 19.562 2.585 34.198 } set { scf_type DF freeze_core True basis def2-tzvpp } energy('sapt2+(3)dmp2') |
结果:
Total SAPT2+(3)dMP2 -158.93980024 [kcal/mol] |
结论
在本文中,用QPCT/SEN177共晶结构PDB 6GBX代理QPCTL/SEN177结合模型,回溯性地重现了Yu等人4QPCTL抑制剂的优化过程。
首先,发现在PDB 6GBX解释的结构中,SEN177存在两种截然不同的结合模式Chain A与Chain B。用apo-GIST对结合口袋进行水分析,初步认为Chain B结合模式是正确的,并可以解释现有的SAR。进一步的holo-GIST分析、MMGBSA结合自由计算以及QM分析相互作用,均支持Chain B结合模式是正确的,而Chain A结合模式是错误的。
分析结合位点,发现两个重要的结合水。然后根据Unhappy water规划了Spark水分子替换策略对SEN177的吡啶母核进行生物等排体替换,重现了经过文献验证过的QP5020设计。这证明了我们的方法能够替换Unhappy桥连水分子并正确的识别出4-腈基苯片段做为吡啶母核的替换结构。
最后,还对三氮唑片段进行生物等排体替换实验,打分最高排名第3的N-甲基咪唑具有最佳的Zn结合几何模式,并被文献报道的QP5038所验证。Spark还命中尚未有文献报道的新化学骨架类型,有望发展出新的系列化合物。
附件
用来重现apo-GIST与holo-GIST计算结果文件可从Github获取:https://github.com/gkxiao/waters/tree/main/PDB-6GBX:
1 2 3 4 | . ├── 6GBX-Chain-A-apo-GIST.flr ├── 6GBX-Chain-A-holo-GIST.flr ├── 6GBX-Chain-B-holo-GIST.flr |
其中FLR文件可以用Cresset的Flare软件打开,我们提供了免费的Flare Visualizer授权: https://www.cresset-group.com/software/licensing-flare
参考文献
- Logtenberg, M.E.W., Jansen, J.H.M., Raaben, M. et al. (2019). Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat Med 25, 612–619. https://doi.org/10.1038/s41591-019-0356-z
- Wu, Z., Weng, L., Zhang, T. et al. (2019). Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRPα-CD47 axis. Cell Res 29, 502–505. https://doi.org/10.1038/s41422-019-0177-0
- Barreira da Silva, R., Leitao, R.M., Pechuan-Jorge, X. et al. (2022). Loss of the intracellular enzyme QPCTL limits chemokine function and reshapes myeloid infiltration to augment tumor immunity. Nat Immunol 23, 568–580. https://doi.org/10.1038/s41590-022-01153-x
- Yu, L., Zhao, P., Sun, Y., Zheng, Z., Du, W., Zhang, L., Li, Y., Xie, L., Xu, S., & Wang, P. (2023). Development of a potent benzonitrile-based inhibitor of glutaminyl-peptide cyclotransferase-like protein (QPCTL) with antitumor efficacy. In Signal Transduction and Targeted Therapy (Vol. 8, Issue 1). Springer Nature. https://doi.org/10.1038/s41392-023-01715-x
- 肖高铿与徐锡明的私人微信通信. 2021年8月25日.
- Spark V10.7. https://www.cresset-group.com/software/spark
- Flare V8.0. https://www.cresset-group.com/software/flare
- Baumgartner, M. P.; Evans, D. A. (2020). Side Chain Virtual Screening of Matched Molecular Pairs: A PDB-Wide and ChEMBL-Wide Analysis. J. Comput. Aided. Mol. Des. 2020, 34 (9), 953–963. https://doi.org/10.1007/s10822-020-00313-1.
- Turney, J.M., Simmonett, A.C., Parrish, R.M., Hohenstein, E.G., Evangelista, F.A., Fermann, J.T., Mintz, B.J., Burns, L.A., Wilke, J.J., Abrams, M.L., Russ, N.J., Leininger, M.L., Janssen, C.L., Seidl, E.T., Allen, W.D., Schaefer, H.F., King, R.A., Valeev, E.F., Sherrill, C.D. and Crawford, T.D. (2012). Psi4: an open-source ab initio electronic structure program. WIREs Comput Mol Sci, 2: 556-565. https://doi.org/10.1002/wcms.93
- PSI4 V1.9. https://psicode.org
- Pozzi, C., Di Pisa, F., Benvenuti, M. et al. (2018). The structure of the human glutaminyl cyclase–SEN177 complex indicates routes for developing new potent inhibitors as possible agents for the treatment of neurological disorders. J Biol Inorg Chem. 23, 1219–1226. https://doi.org/10.1007/s00775-018-1605-1