
摘要:药效团与分子对接技术是常用的两种数据库虚拟筛选方法,那么到底哪种方法更好、还是各有优势呢?中国科学院上海药物所药物发现与设计中心(DDDC)的Zhi Chen等人用Catalyst/Ligandscout、DOCK、GOLD与GLIDE分别针对八个结构多样的靶点进行虚拟筛选、评估、比较了这两种方法的性能,回答了这个问题。
原文:Zhi Chen et al.(2009)
整理:肖高铿
日期:2017-02-01
一. 虚拟筛选的两种常用方法
药效团与分子对接技术是常用的两种数据库虚拟筛选方法,那么到底哪种方法更好、还是各有优势呢?中国科学院上海药物所药物发现与设计中心(DDDC)的Zhi Chen等人用Catalyst/Ligandscout、DOCK、GOLD与GLIDE分别针对八个结构多样的靶点进行虚拟筛选、评估、比较了这两种方法的性能,回答了这个问题。
二. 文献整理
1. 原文出处
Chen, Z., et al. (2009). “Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets.” Acta Pharmacol Sin 30(12): 1694-1708.; doi: 10.1038/aps.2009.159
2. 研究目的
本研究的主要目的是比较两种虚拟筛选方法的性能:基于药效团的虚拟筛选(pharmacophore-based virtual screening,PBVS)与基于对接的虚拟筛选(docking-based virtual screening, DBVS)。
3. 方法
选择八种结构多样的蛋白靶点:血管紧张素转化酶(angiotensin converting enzyme,ACE)、乙酰胆碱酯酶(acetylcholinesterase,AChE)、雄性激素受体(androgen receptor,AR)、 D-丙氨酰-丙氨酸羧肽酶(D-alanyl-D-alanine carboxypeptidase,DacA)、二氢叶酸还原酶(dihydrofolate reductase,DHFR)、雌激素受体-α(estrogen receptors α,ERα)、HIV-1蛋白酶(HIV-1 protease,HIV-pr)以及胸苷激酶(thymidine kinase,TK);分别用不同虚拟筛选方法对它们的活性与decoy化合物进行虚拟筛选(每个靶点分别有两套数据集,都含有活性与decoy数据),然后考察富集因子(Enrichment factor)与命中率(Hit rate)以评估、比较虚拟筛选的性能。
- 富集因子(Enrichment Factor, EF)
- 命中率(Hit rate)
EFx% = (Hitss/Ns) ÷ (Hitst/Nt)
其中Hitss是特定水平下打分靠前的Top x%(比如Top 2%)化合物中含有的活性化合物数;Ns为打分靠前Top x%(比如Top 2%)的化合物数量;Hitst是数据库中活性化合物的总数量;Nt为数据库中化合物的总数量。
Hit Ratex% = 100% × (Hitss/Hitst)
其中Hitss是特定水平下打分靠前的Top x%(比如Top 2%)化合物中含有的活性化合物数;Hitst是数据库中已知活性化合物的总数。
总共测试了四种虚拟筛选方法: 基于药效团的虚拟筛选采用Catalyst软件以及三种分子对接软件DOCK、GOLD与GLIDE,计算流程见图1。
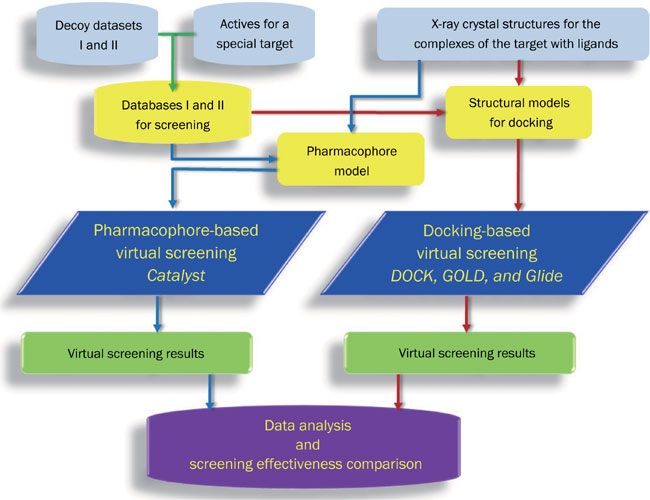
图1. 计算流程(来自原文Figure 1)
其中药效团模型是用Ligandscout采用基于结构的方法从蛋白-配体复合物晶体结构识别而来,图2以ACE为例说明药效团模型的生成过程。
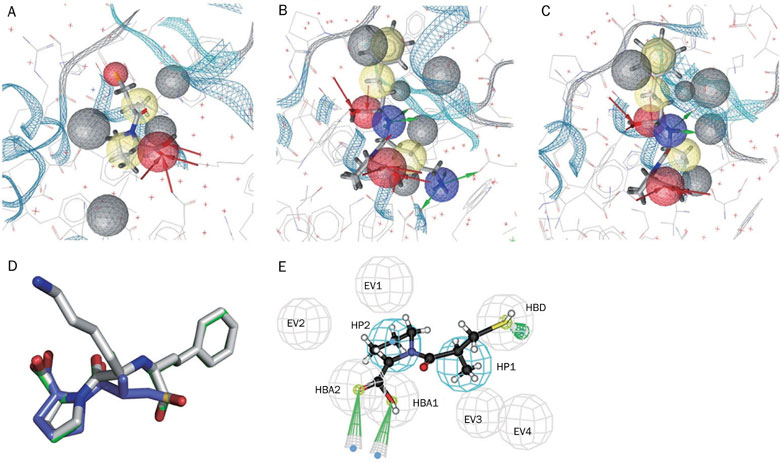
图2. 药效团模型的生成(来自原文Figure 2)。ACE的药效团模型(图A,B,C)是用Ligandscout从ACE-配体复合物结构识别而来(PDB代码分别为1UZF, 1O86, and 1UZE)。药效团元素( pharmacophore features)分别为:氢键供体(hydrogen bond donor,HBD,绿色),氢键受体(hydrogen bond acceptor,HBA,红色箭头), 疏水区(hydrophobic region, HP,黄色球)以及排斥体积(excluded volume,EV,黑色球)。 图D是三个共晶配体叠合到药效团上;图E是用A、B、C药效团模型优化过的药效团模型。最后将药效团模型以Catalyst形式展示: HBA,HBD,HP与EV.
3. 结果
针对16个数据集(每个靶点各有两个测试数据库,分别含有ligand与decoy)进行虚拟筛选之后,比之基于对接的虚拟筛选方法,基于药效团虚拟筛选方法在其中14个数据集中展现了更高的富集因子(见图3)。
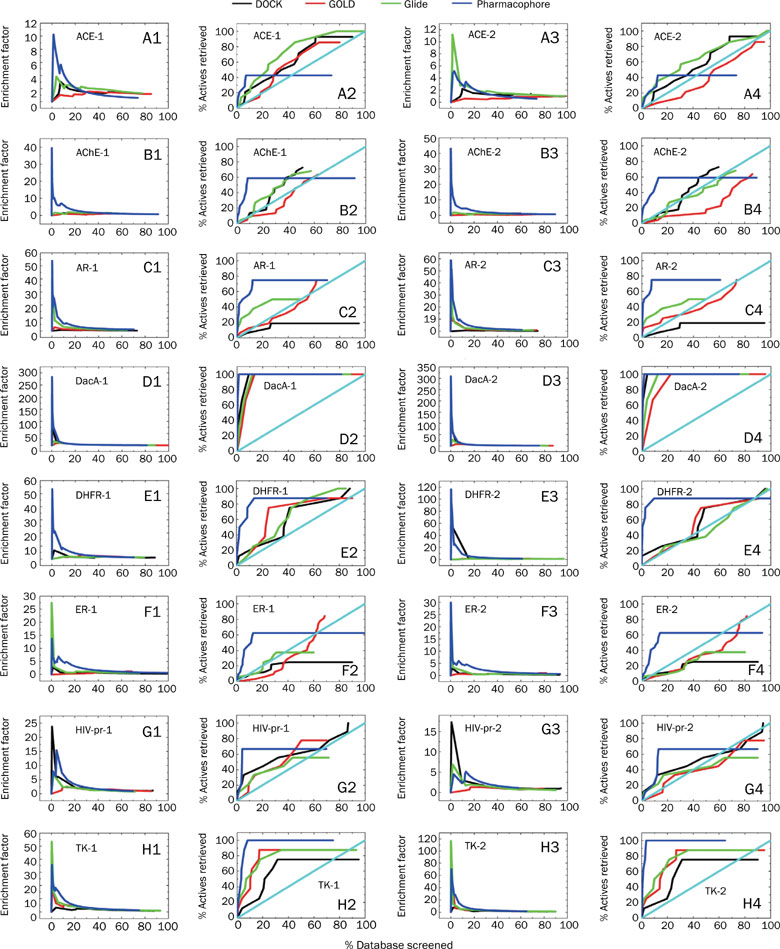
图3.四种虚拟筛选方法对八个不同靶点的虚拟筛选富集因子图(来自原文Figure 3)。富集因子(第1、3列)是数据库按打分、排序后采样比例(Top x%)的函数。随着采集的子集中化合物数(Top x%)的增加,识别出的已知活性化合物数量百分比也增加(第2、4列)。蓝、黑、红、绿色的线条分别代表Catalyst(药效团虚拟筛选)、DOCK、GOLD与GLIDE虚拟筛选结果。-1与-2分别代表数据库I与II。
比之基于分子对接的虚拟筛选,基于药效团的虚拟筛选具有更高的命中率(Top 2%、5%),见图4。
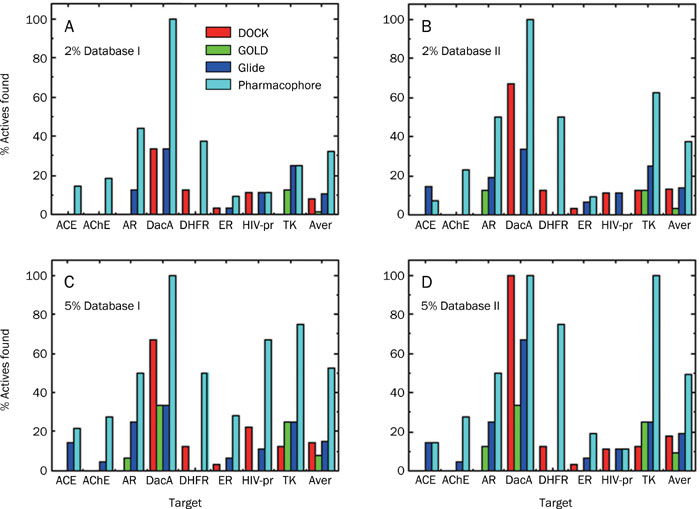
图4. Top2、5%的命中率比较(来自原文Figure 5)。不同虚拟筛选方法命中率(活性化合物百分比)比较:基于药效团(Cyan), DOCK (red), GOLD (green), and Glide (blue)。 2%与5%的意思是取打分最高的2%,5%化合物来考场命中率。最后一列(Aver)为平均命中率。
4. 结论
比之基于对接的虚拟筛选,基于药效团的虚拟筛选在我们的8个测试靶点上表现出更好的虚拟筛选性能,基于药效团的虚拟筛选是药物发现强有力的方法。
三. 讨论
1.基于药效团的虚拟筛选与基于对接的虚拟筛选的比较
分子对接虚拟筛选方法直接反映了配体-受体结合过程,而药效团做为一种查询结构(Query)是配体-受体相互作用的抽象(映射,map)、间接体现了识别过程45。从理论上讲,基于对接的虚拟筛选从数据库中富集活性化合物的性能应该要优于基于药效团的方法。然而事实并非如此,至少本研究的八个靶点虚拟筛选测试中并非如此。我们将这样的结果归因于当前分子对接方法的几个缺陷:
- 第一,基于对接的虚拟筛选采用打分函数来预测数据库中化合物对某一靶点的结合亲合力;
- 第二,大部分现有的对接软件忽略了蛋白的柔性
到目前为止,没有一种打分函数可以通用、精确地针对各种靶点预测化合物的结合亲合力。
这使得在对接过程中出现潜在的活性化合物与现有的蛋白口袋发生碰撞(Crash)而不被命中。
与基于对接的方法不同的是,药效团采用药效团元素(Pharmacophore feature)来表征配体-靶点结合必须的元素,基于药效团的虚拟筛选与排序过程中仅将数据库化合物与药效团进行叠合(拟合)而不考虑实际的配体-靶点结合过程。基于药效团虚拟筛选的这种简化过程实际上将靶点蛋白在结合配体的过程中发生的柔性变化考虑了进来。
将Top 5%的命中活性化合物拟合到药效团与结合位点,观察结果可以证实上述观点。这里我们仅以TK与ERα两个靶点的结果为例来说明为什么基于药效团的虚拟筛选其富集性能优于基于对接的方法。
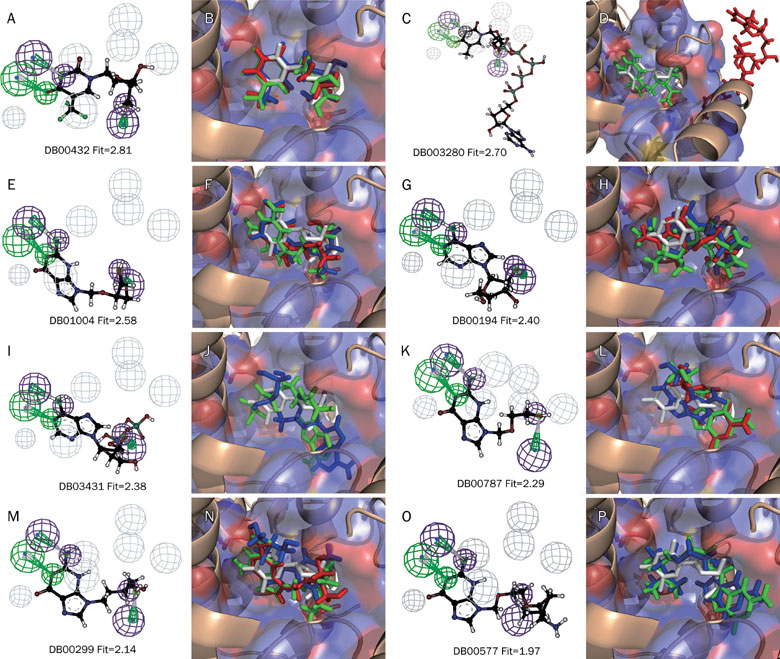
图5.(原文Figure 6) 将活性化合物映射到药效团模型上并拟合到TK的结合位点。Top5%活性化合物是通过基于药效团的方法映射与拟合而得的。药效团模型用Catalyst格式展示(图A, C, E, G, I, K, M,O): 氢键受体 (HBA, 绿色),氢键供体(HBD,紫色), 输水区(HP, 蓝绿色cyan), 排斥体积(excluded volume,EV, 灰色)。 活性化合物的结合构象从虚拟筛选结果中提取出来(图B, D, F, H, J, L, N,P)。其中棍棒状表示的活性化合物来源于对接的结果。共晶结构的配体作为参比展示为白色,Dock、GOLD与GLIDE对接的构象分别用红色、绿色与蓝色表示。
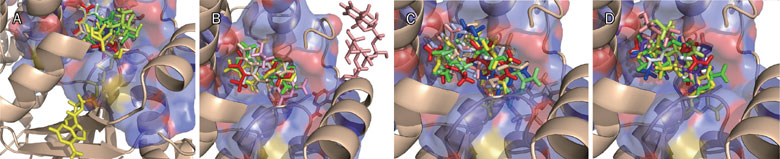
图6.(原文Figure 7) (A) TK活性化合物经药效团映射(Pharmacophore mapping)与结合位点拟合之后的结构叠合图。还展示不同对接方法预测的结合构象:DOCK (图B), GOLD (图C), Glide (图D)

图7.(原文Figure 8) 将活性化合物映射到药效团模型上并拟合到ERα的结合位点。Top5%活性化合物是通过基于药效团的方法映射与拟合而得。图例与Figure 6一致。
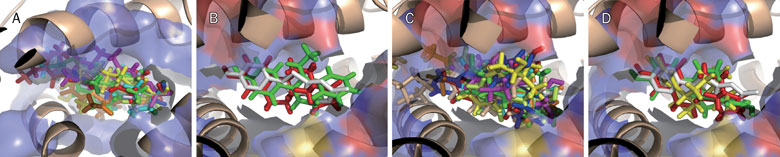
图8.(原文Figure 9) ERα活性化合物经药效团映射(Pharmacophore mapping)与结合位点拟合之后的结构叠合图。图例与Figure 7一致。
对于靶点TK而言, 数据库中总共含有8个已知的活性化合物(Database I与II)。在打分最高的5%的化合物中,基于药效团的虚拟筛选分别从数据库I与II中命中了6个与8个。对接方法DOCK、GOLD与GLIDE仅仅分别命中1、2与3个化合物。TK药效团模型含有3个药效团元素,因此活性化合物与药效团拟合值最佳为3,8个活性化合物有7个与药效团拟合值大于2,仅有一个为1.97(见Figue 6)。这个结果表明,TK的药效团模型是可靠的、虚拟筛选筛选结果也是可信的。然而,对接计算的结果表明:仅有1-2个化合物的可以与TK结合口袋拟合,而其它的化合物不能与口袋互补(见Figure 6右图)。对接方法不能将大尺寸的活性化合物送到结合位点里(见Figure 6 D),或者说对接生成的结合构象远远离开它们的结合构象(见Figure 6J, 6L与6P)。结构叠合也表明:大部分的活性化合物与TK结合口袋的残基发生碰撞(Figure 7)。
对于靶点ERα,基于药效团的虚拟筛选方法从数据库I的Top 5%结果中回收到9个活性化合物。然而,DOCK与GLIDE仅能分别回收到1、2个已知活性化合物,而GOLD没有回收到任何一个已知活性化合物。在TK靶点上也得到相似的结果:全部的9个活性化合物与ERα的药效团模型拟合的很好,拟合值从 2.60到3.74(见Figure 8)。然而,大部分活性化合物不能与ERα结合口袋拟合的很好(见Figure 8、9)。在其它六个靶点上的药效团拟合与对接分析也得到相似的结果。
这些结果表明: 刚性的蛋白晶体结构约束了对接程序从数据库中搜索活性化合物的性能; 基于药效团的方法却不受靶点结构的影响,可以从数据库中通过药效团模型的拟合回收到活性化合物。这意味着基于药效团的方法在虚拟筛选过程中隐式地考虑了靶点蛋白的柔性,这正是为什么基于药效团虚拟筛选方法的性能优于基于对接虚拟筛选方法的原因。
2. 受体柔性是基于药效团虚拟筛选方法性能的一个有利因素
如前所述,基于药效团的方法将化合物拟合到从配体-蛋白复合物结构提取出来的药效团上、并将与药效团拟合的化合物定义为命中化合物(Hit)。Figure 10A展示了化合物DB03431与TK结构的拟合结果。化合物DB03431与1KIM的共晶配体具有相似的骨架,可以与三个药效团元素匹配的很好(Figure 6I):包含有两个氢键供体与一个氢键受体。基于药效团方法预测得到的结合构象与共晶配体(Figure 10A,灰色)相似,可以与残基Gln125发生氢键相互作用,这表明该结合构象是合理的。然而,Catalyst预测的结构构象会与1KIM位于loop区上的G56–G59、K62残基发生碰撞(见Figure 10A浅绿色图)。这个结果表明:基于药效团的方法无需考虑蛋白结构的约束达到容忍更大的立体位阻从而回收更多的活性化合物,而分子对接不会在对接过程中考虑到这种立体效应。比如,GOLD(绿色)与GLIDE(蓝色)对接的结合模式(POSE)显著地偏离了共晶配体(Figure 10A),对接程序DOCK因为立体碰撞而彻底地不能将化合物DB03431送到结合位点。
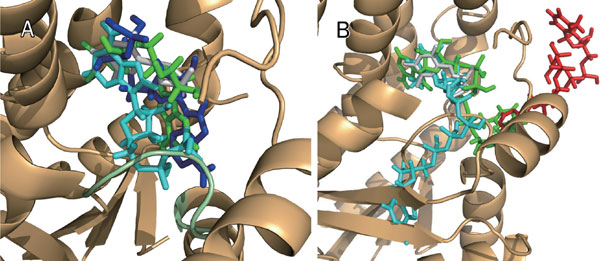
图9.(原文Figure 10) DOCK(红色)、GOLD(绿色)、GLIDE(蓝色)以及Catalyst(青蓝色,Cyan)预测的化合物DB03431(A)、DB03280(B)在蛋白TK结合口袋里的结合构象。
为了进一步阐述基于药效团方法的优势,我们又以DB03280为例进行说明(见Figure 10B)。我们会发现:对接程序处理不了大尺寸的化合物。DOCK与GLIDE未能将化合物送到蛋白的结合位点;GOLD生成一个折叠的、打分很低的构象。与对接方法相反,基于药效团的方法预测到了该化合物的合理构象,该构象与PDB的1KIM共晶配体(见Figure 10B的灰色化合物)相似。尽管预测的构象与蛋白发生碰撞,因为药效团匹配时忽略了残基信息使得基于药效团的虚拟筛(Catalyst)可以将该化合物打分靠前而命中。
四. 相关文章
1. 虚拟筛选假阳性、假阴性产生的原因
参见:http://blog.molcalx.com.cn/2016/07/03/vs-false-negitve-positive-reason.html
2. 虚拟筛选的方法学验证
参见:http://blog.molcalx.com.cn/2016/09/22/virtual-screening-methodology-validation.html


