
摘要:药物化学的优化通常涉对分子的一部分进行替换设计,其目的是在保留化合物活性的同时提高化合物的其他性能。在本研究中,作者进行了回顾性分析:使用多种计算方法从一组随机decoy侧链中识别出活性侧链,从而模仿在实际药物化学项目中可能发生的类似过程。作者通过考察全部的ChEMBL生物活性测试数据(assay),如果一个测试有一个化合物在PDB中以复合物晶体结构出现,那么就收录该assay,从而建立了一个源于ChEMBL与PDB的数据集;此外,还要求该ChEMBL Assay至少有10个测试为活性的化合物并与PDB的那个共晶配体为匹配的分子对;最后得到一个包含402个Assay的化合物数据集。作者用Cresset生物等排替换工具SPARK、Schrodinger分子对接软件Glide以及分子对接软件SMINA等方法对侧链进行打分。在本研究中,作者比较了这些方法将活性侧链从decoy侧链中识别出的性能,并对各种不同方法的使用提出了建议。
原文:Baumgartner, M. P.; Evans, D. A. Side Chain Virtual Screening of Matched Molecular Pairs: A PDB-Wide and ChEMBL-Wide Analysis. J. Comput. Aided. Mol. Des. 2020, 34 (9), 953–963. https://doi.org/10.1007/s10822-020-00313-1.
编译:肖高铿/2020-08-01
前言
在药物开发过程中,人们通常希望对活性分子的整体或部分进行替换以改善其特性(活性,选择性,ADME等)或迁移到新的化学空间以获得结构新颖性。人们已经成功地使用二维与三维配体描述符、蛋白质结合位点信息或两者的组合对现有化合物库进行虚拟筛选发现新的活性分子[1],这已经有相当长的历史了。
本文研究了与虚拟筛选相关但又完全不同的一个问题:在苗头化合物或先导化合物优化项目中,应该去合成什么样的侧链?为此作者创建一个基准测试集,并用了分子虚拟筛选中常用的方法[2– 5]来评估不同侧链替换方法的性能。
替换一个分子的一部分而保留其余部分不变会得到一个匹配的分子对(Matched molecular pair ,MMP)。 将相似的化合物组成MMP或将几组化合物组成MMP系列,已经成为一种流行的理解、预测化合物SAR的有力方法[6,7]。MMP有多种不同的形式,在本研究中,将MMP定义为可以通过断开一个非环单键来切换侧链的化合物。
在本研究中,作者从公开的结合亲和力和结构信息中生成了一系列MMP系列的数据集,并测试了多种基于结构和基于配体的方法,以确定每一种方法从一系列decoy侧链替换中正确识别出活性侧链的良好程度。每个数据集包含10个或更多在同一ChEMBL assay中被证实具有活性的MMP,要求在每个assay中至少有一个化合物已有相关蛋白质的共结晶PDB。
方法
数据集的注释
作者按照Figure 1概述的流程构建了数据集。由于要对基于结构的方法进行测试,因此不仅使用了结合亲和力数据而且使用了结构生物学的数据来编译数据集。数据集是通过搜索ChEMBL 20 [8-10]中所有生物活性测试(assay)而生成的,其中活性测试的蛋白也必须与活性测试中一个化合物结合并以复合物出现在PDB。对10,672个匹配的测试进一步过滤,筛选出其中至少有10个化合物在该测试中表现出活性并与PDB中的配体成MMP的那些测试。作者将MMP中的公共部分称为“context”,将变化部分称为“侧链”(见Figure 1)。用KNIME节点“Automated Matched Pairs” 来识别每个测试中的MMP [11-14]。在本研究中,作者将活性化合物定义为:测定亲和力在PDB配体的0.5 log单位(pIC50,pKi等)以内或更大范围内的配体。而且还要求侧链至少具有六个重原子,以消除细小的变化(即甲基化)。经过这些过滤器后,剩下402个数据集,包含四个标识符:ChEMBL Assay ID,PDB ID,共晶化合物的ChEMBL ID,以及MMP的公共子结构SMILES。数据集中的活性化合物的数量从10到67不等(平均值:15.5±6.6)。补充信息提供了ChEMBL assay ID、化合物 ID、以及准备好可以用于对接的PDB结构。

Figure 1. 数据集的注释流程。以MMP13为例,PDB 4A78的配体在ID号为CHEMBL1943704的CHEMBL Assay中进行过活性测试。
Decoy侧链
作者还准备一组Decoy侧链,在虚拟筛选中用作假定的阴性数据。作者从ChEMBL 20[8-10]中随机选择了1000个生物活性测试实验(Assay)。对于每个测试,提取出所有可能的MMP;仅保留至少包含六个重原子的已识别的侧链,过滤掉其中重复的侧链,最后获得了包含7559个decoy侧链的数据集。
对于每个数据集中的活性化合物,用基于与活性化合物理化性质相似性的概率加权法选择了50个decoy侧链。理化性质是使用RDKit[8]计算:SlogP,分子量,可旋转键数量,氢键供体数量,氢键受体数量和净电荷。用概率f(di || a)来选择Decoy侧链di,其中f(di || a)是活性化合物理化性质的多元正态分布,均值以活性化合物的上述性质的值为中心,协方差矩阵的对角线设置为(0.5、30、1.0、1.0、1.0、0.1),所有非对角线值设置为零,每个活性化合物最多选择50个decoy侧链,并去除重复结构。此外,还需要将已知活性化合物从Decoy中剔除。Decoy选择过程的灵感来自于全分子虚拟筛选DUDE decoy构建方法[15]。
打分方法
作者评估了7种方法区分已知活性侧链与decoy侧链的能力。在Aign-Minmize方法中,化合物的构象用RDKit[16]产生,并用RDKit将化合物叠合到共晶配体的公共结构上,然后用Smina[17,18]在PDB受体蛋白背景下进行结构能量最小化。在Smina分子对接方法中,SMINA用默认参数并以共晶配体为盒子中心将化合物对接到蛋白晶体结构,打分最靠前pose的分值用于化合物排序。在Glide SP[9-12]对接方法中,Glide SP软件采用默认参数并以共晶配体为格点中心将化合物对接到蛋白晶体结构,化合物打分最靠前的那个pose用于排序。所有方法所用的蛋白结构的输入文件与共晶配体的结构都是用Schrodinger的蛋白准备流程(protein preparation protocol)在默认参数下准备出来的:加氢、删除配体5Å之外溶剂分子、用OPLS3.0力场对结构进行优化并限定重原子相对于输入坐标最大RMSD为0.3[21,22]。所有准备好的蛋白与配体见支持信息。
还用Cresset的SPARK[23,24]对化合物进行了排序。用SPARK在默认参数下对活性与decoy侧链都进行了碎片化。SPARK搜索时采用PDB共晶配体的pose作为起始分子,搜索参数如下:shape weight (-s) = 0.5, maximum heavy atoms (− n) = 35, and maximum results (− m) = 5500。
还测试了上述SPARK方法的两种衍生方法。SPARK+排斥体积方法,除了使用蛋白结构作为排斥体积之外,该方法与SPARK方法完全一致。SPARK Lig方法与SPARK一致,除了它不是用共晶配体的pose作为起始分子之外,而是用Schroginer/ConfGen[25]在高通量模式下生成一个最低能构象,除此之外其它都与SPARK方法一致。
最后一个方法是计算活性与decoy侧链与共晶配体侧链的2D相似性。RDKit指纹[16],与Daylight指纹[26]相似,用于计算Tanimoto相似性。然后用计算的侧链与PDB共晶配体侧链的Tanimoto相似性进行排序。
性能的打分标准是ROC AUC。ROC AUC取值范围从0-1,1.0意味着性能完美,0.5意味着性能与随机一样[2]。还用BEDROC(20)作为早期富集[2]能力的打分标准。
结果
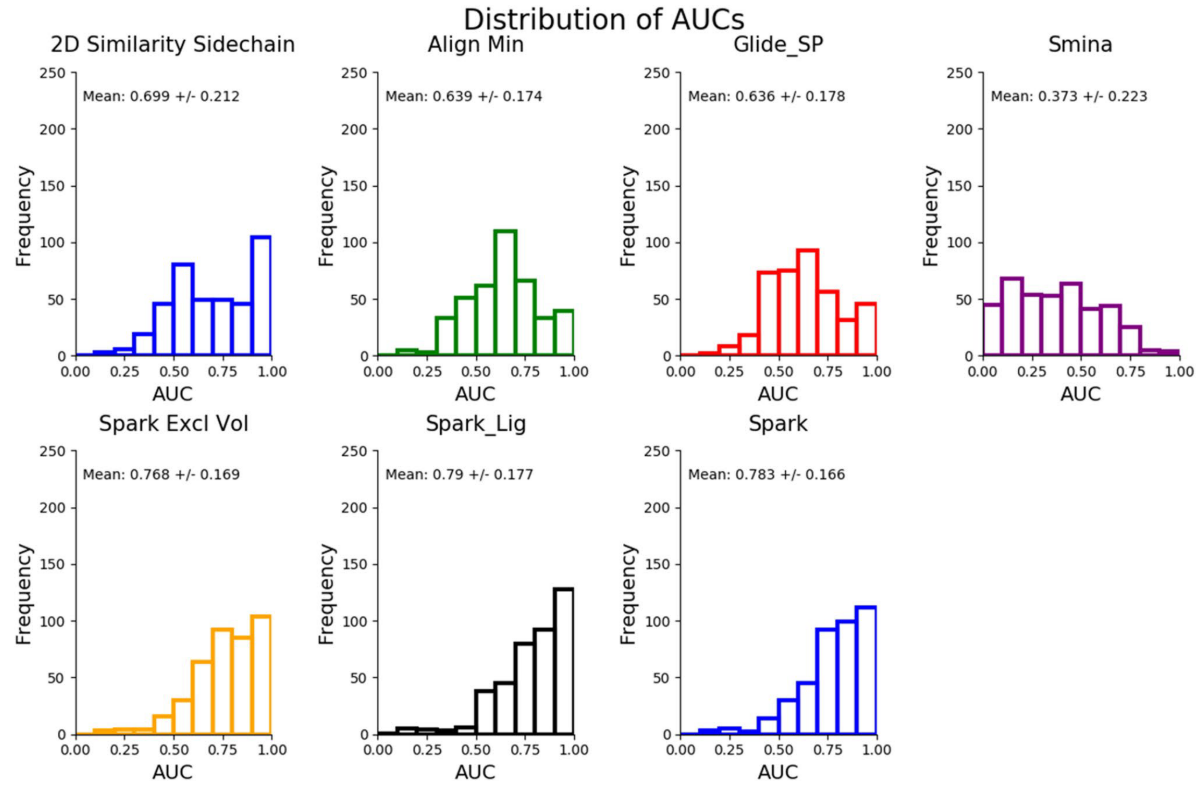
Figure 2. 7种方法在402个数据集上的AUC总体分布
6种方法都用402个数据集进行了测试,并计算了ROC AUC以评估这些方法区分活性侧链与Decoy侧链的能力。Figure 2展示了每种方法AUC的总体分布。总的来说,SPARK方法在一系列数据集上效果很好。
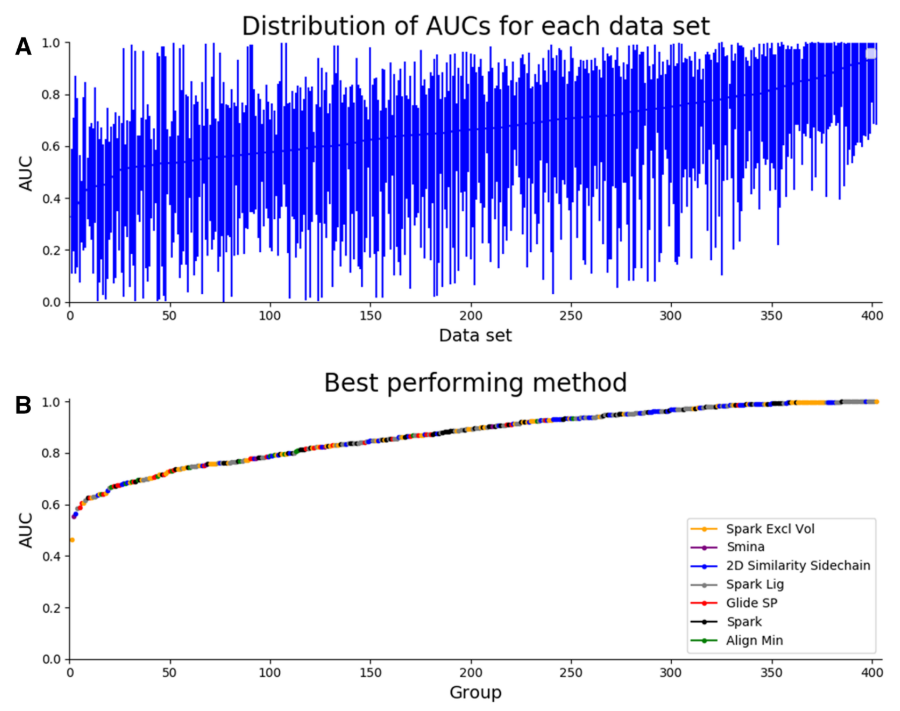
Figure 3. a:每个数据集的AUC值的范围; b:每个数据集的最佳AUC及其对应方法
作者研究了这些方法在每个数据集上的性能表现以了解是否某些数据集对所有的方法都难或都容易。Figure 3a给出了这些方法对每个数据集的最小、最大性能以及平均性能。结果表明,有的数据集对所有的方法都很容易(高的最小AUC,Figure 3a越往右越容易),有的数据集对所有方法都难(低的最大AUC,Figure 3b越往左越难)。Figure 3b展示了对各个数据集性能最佳的方法。
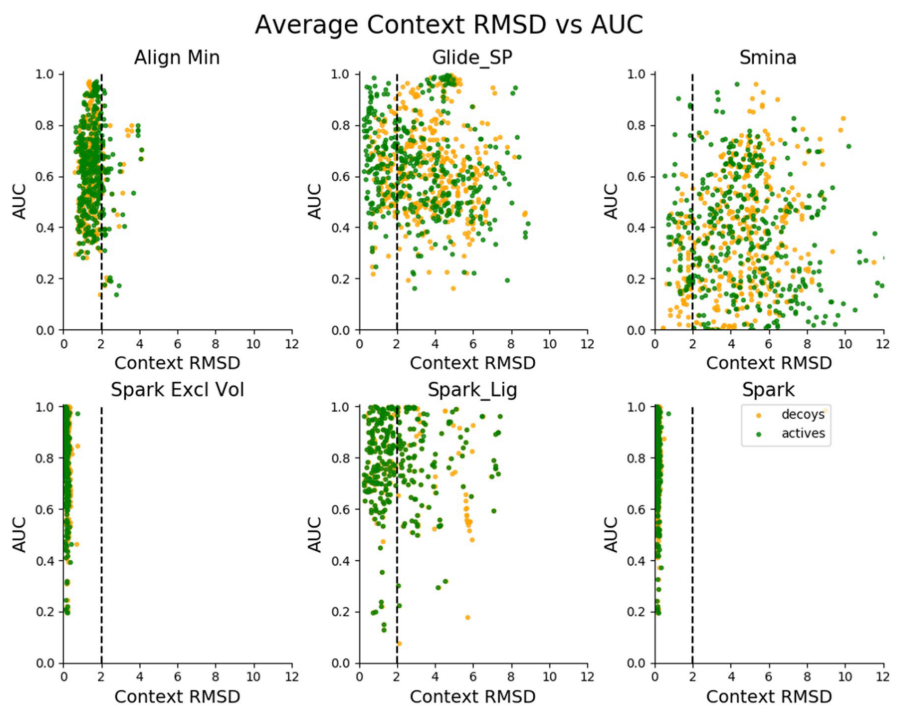
Figure 4.活性(绿色)与decoy(黄色)化合物公共子结构的平均RMSD与AUC
作者计算了各种方法打分化合物的公共结构部分的RMSD以确认是否pose预测差会导致对活性化合物的打分差。化合物公共结构的RMSD是用OpenBabel[27]的obrms计算的。Figure 4比较了活性化合物与decoy化合物的公共结构平均RMSD与每种方法的AUC。预计会看到,基于结构方法在差的pose预测性能与差的虚拟筛选性能之间存在着相关性。如Figure 4所示,在某种程度上,在Smina方法上确实看到这样的相关性:pose的平均RMSD越高,AUC也趋向于越低,虽然相关性并不强。有很多数据集pose预测的很差(RMSD大于等于2 Å),但是却有着相当好的虚拟筛选性能(AUC大于等于0.8)。然而,在将AutoDock Vina打分函数与Align Min方法进行比较时,确实观察到叠合得到pose用AutoDock打分函数进行最小化后可以看到AUC的改善(Figure 2),这与pose预测得到改善是一致的(Figure 4)。
SPARK与SPARK+Excluded Volume这两个方法都保留着公共结构部分在空间中不变而仅仅添加了侧链,所以这两个方法的公共子结构部分RMSD≈ 0 Å。

Figure 5.PDB共晶配体侧链SASA与AUC的比较。对于每种方法,都会计算PDB配体侧链的SASA,并将其与该数据集的AUC进行比较。
为了确定替换后的侧链埋在口袋中或暴露于溶剂中的数据集之间是否存在打分性能差异,并由此导致基于结构的方法可能会由于缺乏与蛋白质的接触而遇到困难,作者计算了 PDB配体侧链的平均重原子溶剂可及表面积(SASA),如Figure 5所示。其中共结晶配体侧链的SASA使用asacalc工具计算而得[28]。
可以预计,由于缺乏与蛋白质的接触,因此侧链暴露于溶剂的数据集采用基于结构的方法将更加困难。 但是,诸如Glide和Smina的对接方法却没有表现出很强的相关性。 这表明侧链的溶剂可及性不是这些数据集打分性能的主要决定因素。
失败的算例
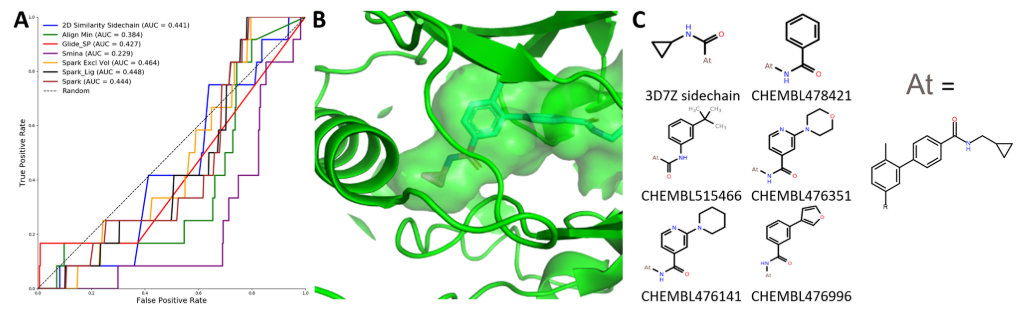
Figure 6. a: 所有方法在PDB 3D7Z配体(CHEMBL assay ID CHEMBL987298)数据集的ROC曲线;b:PDB 3D7Z结构可以观察到被包埋的侧链;c:一些活性化合物的侧链例子
作者考察了几个多种方法或所有方法均失败的算例。其中一个算例是对PDB 3D7Z共晶配体侧链进行打分(ChEMBL assay ID CHEMBL987298),各种方法的平均AUC为0.405,最佳方法是SPARK Excl Vol(AUC=0.464,差于随机,见Figure 6a)。由于多种潜在原因,该数据集被证明特别具有挑战性。作者认为,在该算例中,由于与侧链相互作用的口袋很小,所以基于结构的方法难以解决。Figure 6b显示了共晶配体的侧链以及与之结合的受约束的结合口袋。Figure 6c显示了PDB配体的侧链结构和活性侧链替代物。其它的活性侧链显著地大于共晶配体的侧链,基于结构的对接方法如果不对蛋白侧链进行采样重排的话,则这些较大的侧链将与结合位点不匹配。由于查询化合物(编者注:参比化合物)和活性侧链之间具有较大的尺寸和化学差异,所以SPARK可能难以正确地对活性化合物进行打分。
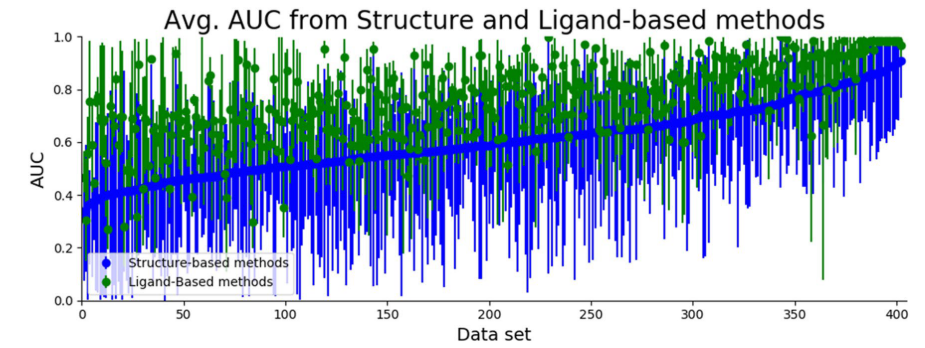
Figure 7. 基于结构(蓝)与基于配体(绿色)方法对每个数据集的平均性能,其中数据集按基于结构方法平均AUC进行排序。
在本研究中,基于配体的方法通常优于基于结构的方法。如 图7所示,基于配体的方法平均性能优于基于结构的方法。假设这再次可能是由于对接方法(见Figure 4中的Glide_SP和Smina)总体pose预测较差所致。
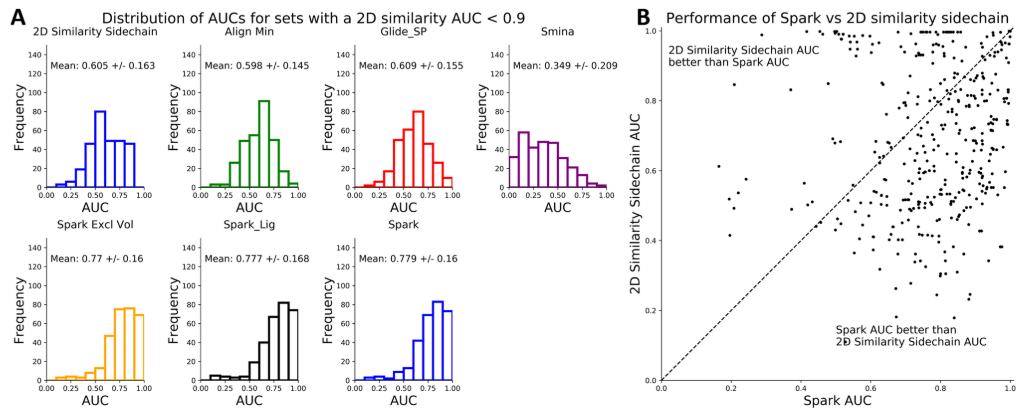
Figure 8. a:2D相似性小于0.9数据集AUC分布;b:SPARK与2D相似性方法的性能比较,虚线型对角线下、上方的点分别对应于SPARK识别活性化合物性能优于、差于2D相似性的数据集分布。
除了基于结构和配体的方法之外,作者还测试了基于与PDB配体2D相似性对侧链打分的方法。如Figure 1所示,在区分活性化合物和decoy化合物方面,2D相似性方法在几个数据集上表现很好(AUC大于等于0.9)。在这些数据集中,简单地计算与苗头化合物(PDB共晶配体)的2D相似性就非常有效,这表明PDB共晶配体和活性化合物结构非常相似,仅因微小变化而不同。作者认为这些数据集不那么令人感兴趣,因为它们仅代表了典型药物化学团队常规所作的结构替换。为了确定其它方法是否仅由于大多数活性成分与已知苗头化合物具有明显(2D)相似性而表现良好,作者将仅使用2D相似性就可获得良好打分的数据集剔除掉(Figure 8a)。
删除这些难度低的数据集不会对方法性能的分布产生很大影响。SPARK方法生成的AUC分布基本上没有变化。其他方法Glide_SP,Smina和Align Min的平均性能略有下降。
SPARK(不使用排斥体积)是一种基于配体的方法,该方法通过计算与查询分子(参比分子)的形状和静电相似性而对生物等排体(替换结构)进行排序。Figure 8b显示了SPARK与2D相似性方法在每个数据集上的性能比较。可以看到,SPARK产生的场相似性(Field similarity)比单独使用2D相似性具有更好的预测性能。
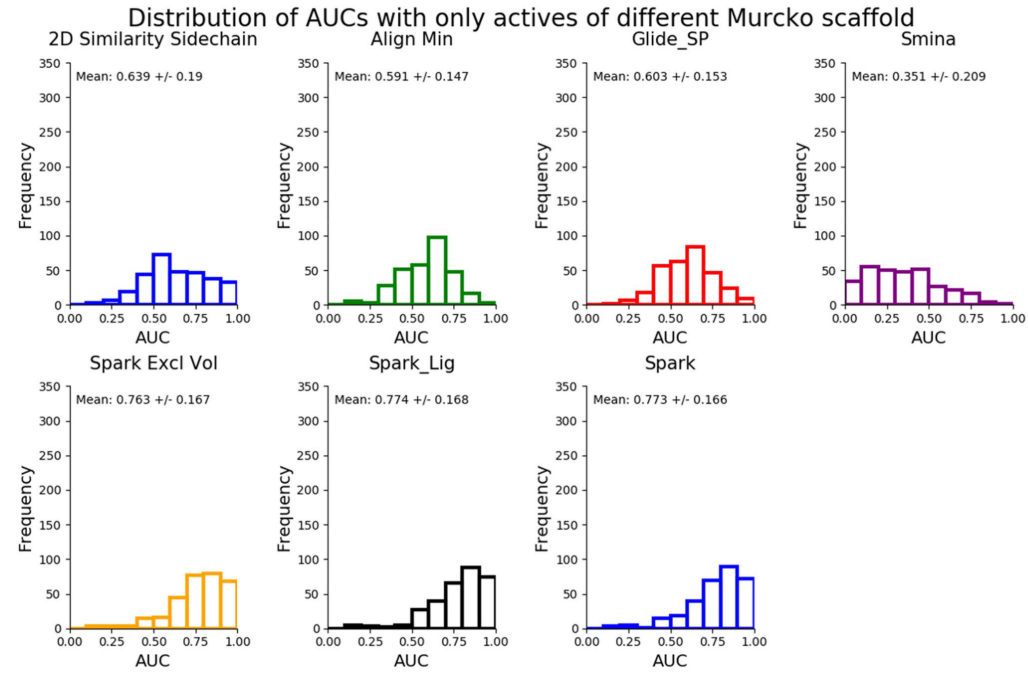
Figure 9. 当仅考察具有不同马库什骨架的活性化合物时,6种方法的AUC分布
作者还考察了这些方法骨架跃迁的能力,即识别与苗头化合物(共晶配体)具有大的化学结构变化的其他活性侧链的能力。作者计算了PDB配体侧链马库什骨架(Murcko Scaffold)[29]和活性化合物侧链的马库什骨架,并将具有相同马库什骨架的活性化合物剔除后重新计算了AUC。由于有些数据集仅包含来自同一马库什骨架的活性侧链,因此这些数据集被排除在本次分析之外。用剩余的286个数据集重新计算了AUC,其分布如Figure 9所示。总体而言,性能没有发生显著变化,这可能是因为效果良好的方法对不同的骨架具有很高的打分。
不同类型靶标的性能差异考察
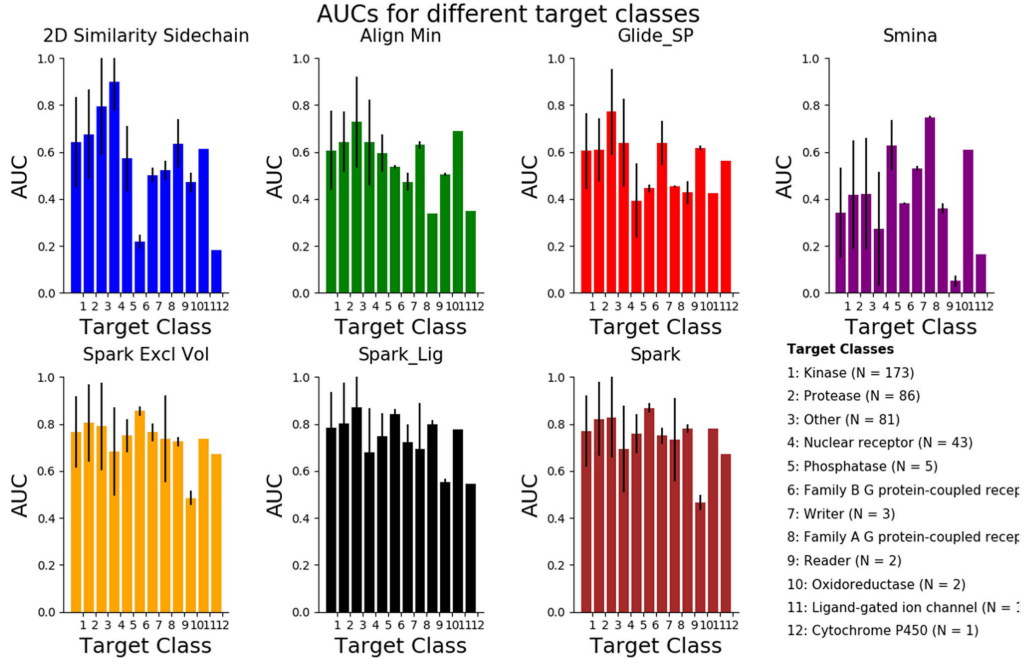
Figure 10. 不同方法在不同靶标上性能表现
作者还考察了各种方法对不同靶标类型的性能。对于ChEMBL assay中列出的每个蛋白靶标,作者绘制了这些方法对每个靶标类别的性能图。 可以观察到,靶标类别之间的性能差异很小(Figure 10)。请注意,每个靶标类别中靶标的数量高度不均匀,这反映了历史上科学和制药工业对该靶标类别的兴趣度以及该靶标结构解释的难易度。事实上,本研究数据集中的大多数靶标是激酶和蛋白酶。注意到Figure 10中1-4类靶标(激酶,蛋白酶,其他和核受体)占了数据集的大部分,可以看出来:每种方法的性能通常是一致的。由于不太常见的靶标的数据集数量很少(N = 1-5),因此无法评估这些方法对这类靶标的性能。
讨论与结论
在本研中,作者构建了一个数据集,用于对许多侧链虚拟筛选方法的虚拟筛选性能进行基准测试。该数据集包含了402个子集,由源自ChEMBL assay的活性化合物组成,每个子集至少有一个化合物为PDB共晶配体并至少包含10对活性化合物匹配对(MMP)。
作者测试了多种基于结构和基于配体的方法,以考察这些方法区分已知的活性侧链和decoy侧链的能力。作者观测了这些方法和数据集的性能表现。在本研究中,有许多数据集可以被任意的虚拟筛选方法可靠地识别出活性侧链,因此这样的数据集似乎更容易被预测(Figure 3a,左侧)。 相反,也有一些数据集,所有被评估的方法均无法识别出其中的活性侧链(Figure 3a,右侧)。
作者分析了数据集的特性,以试图确定哪些蛋白或配体的特性可以暗示性能最佳的虚拟筛选方法。发现差的pose预测与基于结构的方法区分(活性侧链与decoy侧链的)能力之间只有微弱的相关性(Figure 4),但请注意,使用根据参比分子(共晶配体)叠合获得pose的Align Min方法比起用同样打分函数(编著注:同样的打分函数是因为在Align min方法中也用Smina在结合位点中对配体进行minimization)但经常对接不对的Smina方法相比,前者比后者的总体性能好得多(Figure 2)。 作者还发现,增加侧链溶剂可及性会导致对接方法的性能略有下降,但影响很小。
总之,SPARK从大范围靶标、多种化学类型的Decoy中识别出活性匹配分子对侧链的性能表现良好(402个数据集的平均AUC=0.783)。作者还假设,在本研究中基于对接的方法往往会遇到困难,这是由于受到力场的限制:力场似乎缺乏区分同类系物相对较小结构差异的能力。
本研究的一个主要局限是评估方法没有考虑蛋白的柔性。对接方法(Glide和Smina)和SPARK仅使用蛋白结构的一个构象进行预测,多个案例已经证明这一假设是错误的[30]。作者限于使用这些方法有两个原因。首先,通过分子动力学模拟、系综对接或其它将蛋白柔性考虑在内的方法在计算上是昂贵的,并且通常不能很好地扩展到大量化合物的应用上。因此,作者聚焦于适用于筛选的高通量方法,例如筛选适于特定反应的、有库存或可购买的所有可用化学试剂,其数量通常为数万或更多。其次,作者认为,有限的结构信息(一个或只有几个晶体结构)代表了学术界和制药工业界在项目开始时的典型情况,希望本评估具有尽可能广的适用性。
在未来的工作中,了解诸如如自由能微扰(free-energy perturbation,FEP)[31]之类计算更昂贵、精度更高的方法如何增加侧链虚拟筛选或类似虚拟筛选的价值,将是有益的。
总之,从公共蛋白-配体活性数据(ChEMBL)与结构生物学数据(PDB)交集中采集了匹配分子系列,构建了基准数据集。对多种当前最先进的方法进行了虚拟筛选测试,以评估这些方法从decoy侧链中识别出活性侧链的能力。尽管所评估方法的准确度水平一般很高,但显然在对匹配分子系列进行排序时仍有很大的改进空间。
文献
见原文
评论
本文作者建立了一套评估R-基团虚拟筛选的数据集,然后对SPARK进行了全面(用不同的设置)的测试,并与2D相似性、RDKit叠合然后Smina打分、Smina对接、Glide对接进行了比较。结果表明,SPARK性能最佳。
重要的几点引述如下:
“In general, the Spark methods worked well across a range of data sets.”
“Figure 8b shows the performance of Spark compared to the performance of the 2D similarity for each data set. We can see that the field similarity produced by Spark is adding predictive value over using the 2D similarity alone.”
“Removing these less challenging datasets did not have a large effect on the distributions of the performance of the methods. The distributions of AUCs produced by the Spark methods were essentially unchanged. ”
“Overall, we found that Spark performed well at identifying active matched pair side chains from a pool of decoys across a wide range of targets and chemotypes (average AUC = 0.783 for the 402 datasets ”
相关主题
- 肖高铿. 十种分子对接软件的性能评估
- 肖高铿. 如何进行虚拟筛选的方法学验证
- 肖高铿. FLARE算例 | 炼金术法FEP预测FXR激动剂的结合自由能
这篇博客文章编译自浙江大学侯廷军老师课题组的文章,该文章考察了10个分子对接方法的虚拟筛选性能、结合模式预测性能与打分性能。
该博客文章介绍了虚拟筛选方法验证的一些重要指标计算方法。
D3R GC2竞赛的Free Energy Set2包含了18个活性跨度达3个数量级(IC50差异达1000倍)的同系物,其中FXR_12有PDB复合物结构可以下载,该数据集完全满足本文侧链虚拟筛选Benchmark的定义,且具有一定的挑战性。结果表明,FEP非常完美地将活性高与活性低化合物区分开了。


