
摘要:炼金术自由能计算是一种有用的工具,可用来预测与分子从一种环境转移到另一种环境时的自由能差异。该方法的标志是使用“桥接”势能函数,该函数代表了不能作为真实化学物种存在的炼金中间态。从这些桥接的炼金术热力学状态收集的数据允许有效地计算自由转移能量(或转移自由能量之差),而模拟时间要比直接模拟转移过程少几个数量级。尽管该方法很灵活性,但必须注意避免常见的陷阱以确保计算出的自由能差对于选定的力场而言是可靠的且可重现的,并且应包括适当的校正以允许与实验数据直接比较。在本文中,作者回顾了炼金术自由能计算几个流行应用领域的当前最佳实践,包括小分子与生物靶标间的相对和绝对结合自由能计算。
作者:硅康SiliconTX
声明:经硅康医药(苏州)有限公司徐华锋老师许可转载
1 什么是炼金术自由能计算方法
炼金术自由能计算可计算与转移过程相关的自由能差,例如,小分子与受体的结合过程或小分子从水相到非极性相的转移过程。这些计算使用许多非物理状态的中间状态,其中系统的某些部分(例如小分子配体或受体侧链)的化学同一性通过修改被修饰、插入或删除的原子间相互作用的势能函数来改变。图1说明了常见的自由能变化,这些变化可能很难用常规计算方法来计算,但更容易用炼金术方法计算。
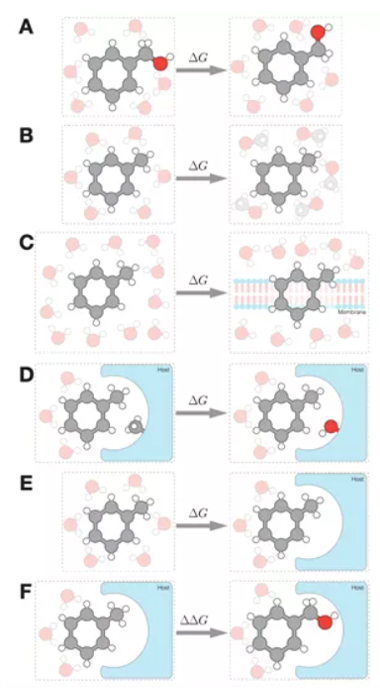
图1: 可以利用炼金术自由能方法计算的几种常见的自由能变化过程: A)由分子构象变化引起的自由能变化;B)配分系数,例如 log P 或者 log D;C)分子插入膜蛋白过程的自由能变化;D)由蛋白或宿主残基的突变引起的结合自由能变化;E)小分子与蛋白或者宿主的绝对结合自由能变化;F)一个分子和另一个分子与同一个靶标蛋白的相对结合自由能变化。
2 先决条件与应用范围
本指南旨在回答以下问题:
- 所研究课题是否可以选择炼金术自由能计算?
- 如何选择正确的炼金术自由能模拟协议并运行它?
- 如何准确地分析炼金术自由能模拟?
- 哪些软件工具可用于执行炼金术自由能计算?
3 炼金术自由能计算的统计力学
假设计算配体L与蛋白P的结合自由能:
$$
P + L <=> PL
$$
其平衡结合速率可以由产物[PL]和反应物[P]与[L]的浓度之比而得到:
$$
K^{o}_{b} = c^{o}\frac{PL}{[L][P]}
$$
为了定义参考态,标准态通常被定义为1 mol/L,c◦表示标准态浓度。因此,结合自由能可以由下式得到:
$$
\Delta G_{bind} = RTlnK^{o}_{b}
$$
我们可以利用配分函数之比来计算分子体系在结合态和未结合态中出现的概率,得到结合速率:
$$
\Delta G_{bind} = RTln\frac{\int_{bound}exp(-{\beta}U(x))dx}{\int_{unbound}exp(-{\beta}U(x))dx}
$$
或者,更一般简化结合态与未结合态之间的自由能差的计算形式:
$$
\Delta f \equiv f(bound) – f(unbound) = -ln\frac{Z(unbound)}{Z(bound)}
$$
在这里,
$$
Z=\int_{bound}exp(-{\beta}U(x))dx
$$
代表构象积分。我们很难直接模拟计算得到结合态与未结合态的构象积分。此时,炼金术自由能模拟就很有必要了。如图2所示,利用该图中的热力学循环,我们可以计算得到两个配体A和B之间的相对结合自由能。
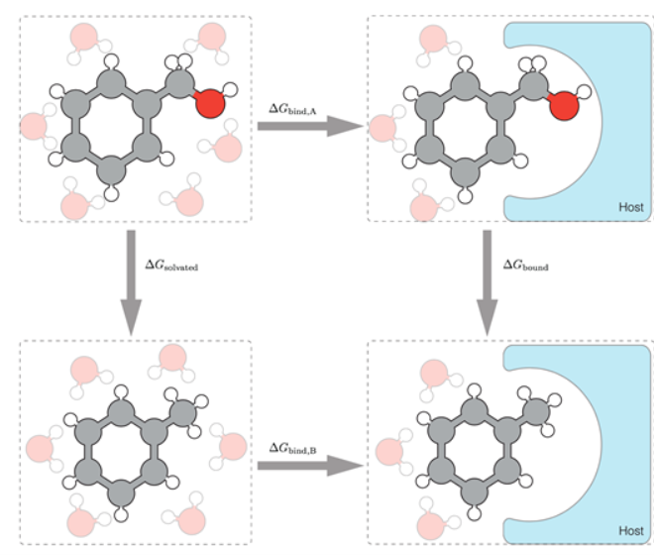
图2: 热力学循环—计算两个小分子之间的相对结合自由能 ∆∆G
炼金术自由能计算的定义特征:利用一系列修改的势能函数U(x;λ)来做模拟。其中,参数λ可以调控在真实化学体系中不会出现的相互作用。用一个或者多个模拟来收集所有热力学态的能量以计算初态(λ0)和末态(λ1)之间的自由能差:
$$
\Delta f \equiv f(\lambda _{1}) – f(\lambda _{0}) = -ln\frac{Z(\lambda _{1})}{Z(\lambda _{0})}
$$
4 从炼金术模拟中可以期待什么
4.1 炼金术自由能计算的准确度如何
在可行的例子中,目前的炼金术自由能计算似乎可以实现1-2 kcal/mol左右的RMS误差,具体取决于力场、体系和各种其它因素,例如模拟时间、采样方法以及所采用的计算是否是绝对的或相对的。特别地,相对的计算通常需要结构上比较相似的配体作为起点。诸如蛋白质或配体重排的长时间尺度,配体结合模式的不确定性或带电荷的配体之类的其它因素会使这些计算的可靠性大大降低,需要更多的研究工作。值得注意的是,自由能计算的准确性在不同的蛋白质靶标之间变化很大,在不同的配体化学型之间也可能存在很大差异。
4.2 炼金术自由能计算的可重复性如何
有限的计算资源必然会限制对势能面采样的不相关样本的数量,因此,炼金术自由能计算给出的自由能值会限制在有限的精度范围内。
在简单的体系中,例如有机小分子的绝对水合自由能,或结构相似的有机小分子之间的相对水合自由能,应该可以使用给定的软件包获得高精度的估算值(标准偏差在0.01 kcal/mol以下)。对于更复杂的体系,例如蛋白-配体结合自由能,可重复性通常会大大降低。一个好的做法是重复两次或三次相同的计算,以评估既定模拟协议的可重复性。
请注意,模拟程序包的变化也会带来其它问题。由于方法上的差异,例如积分器、恒温器、恒压器、远程静电的处理以及潜在的其它因素,可重复性方面会有更大的可变性。
4.3 炼金术自由能计算的适用性如何
当需要(缓慢或昂贵的)合成才能进行实验或实验成本过高时,自由能计算才有吸引力。而且,自由能计算可以给出实验难以得到的答案。
4.4 炼金术自由能计算的准确度是否足够
如果是为了在合成许多化合物时优化先导化合物,那么即使在1–2 kcal/mol的范围内精确地计算自由能也是很有吸引力的,但是如果要合成的化合物数量非常少,这种精确性可能不足以提供很多价值。
4.5 炼金术自由能计算模拟的成本如何
关于“成本”,与实验相比,不仅指的是计算所产生的花费成本,还有时间成本,即计算是否快于实验。
4.6 是否需要探索性研究
如果不确定所研究项目是否可行,一个可能的选择是先进行一个简短的探索性研究,以评估为数不多的配体的可行性。这足以初步了解所提出目标计算的可行性和准确性。
5 炼金术自由能计算模拟应该如何应用于药物研发项目
5.1 考虑实验条件
有必要考虑实验条件设定的细节,例如pH值。更详细地说,小分子的电荷或互变异构态可以在一系列类似物中改变。另外,为了保证模拟模型与实验相匹配,我们需要保证是对同一个体系进行模拟。此外,在生物测定中是否需要考虑某些共同因子或者伴侣蛋白。
5.2 结合模式是否准确
一个较好的炼金术自由能计算要求准确的配体结合模式,这通常是基于相关配体的X射线晶体学来估计的。配体和蛋白质在与相关配体的结合方式上也会发生意想不到的变化,从而使这些问题更加复杂。
5.3 输入设定与计算规模
考虑几十个(或更多)配体是很常见的,因此,有必要在结合位点处将它们进行叠加排列。目前还没有深入研究不同的叠加排列方法是如何影响计算结果的,但使用者应注意这些实际的考虑。
目前,水分子在配体结合中的作用已经被很好地理解。在配体结合过程中捕捉结合位点溶剂化的变化是至关重要的。
在药物研发程序中,结合自由能计算通常需要使用软件或工具来加速。
5.4 预测、理解错误
在设计合成化合物以了解预测中潜在的误差或不确定性时,了解可能的误差并考虑选择偏差是非常重要的。
回顾性评估可以为类似分子的预期性能提供指示。几个参数提供了有用的性能指标。结果表明,对于相对结合自由能计算,MBAR误差大于0.3 kcal/mol可能是不准确的指示。此外,可以通过比较相同结构变化的正向和反向自由能计算的差异来检查迟滞差异,或者在变化较大的情况下,循环闭合误差表明连接许多化合物的循环可能涉及有问题的自由能计算。选择偏差在未来的应用中可能会有问题,因为通常的目标是只关注一个较为狭窄的活性预测范围。
从药物研发的角度来看,自由能微扰正在扩展其应用领域。并且有研究发现,该方法已经成功地使用了同源模型和GPCRs,以及实现了电荷变化和骨架跃迁,但这些体系研究无疑更加困难。同时,应用实例正在扩展到抗性预测、疾病突变预测、溶解度预测——这些都是炼金术自由能计算的光明应用前景。
6 自由能计算模拟前提条件
原则上,在足够充分的构象采样的极限情况下,由炼金术自由能计算给出的自由能差应该是与体系的初始结构坐标无关的。然而,在实际情况下,由于热力学态模拟时间的有限性(通常在1到100纳秒的尺度),我们需要考虑以下几个方面来选取初始结构坐标以获得满意的计算结果:
- 是否可以得到足够好的 X-ray 结构?
- 如何处理不同的结合模式?
- 是否考虑了立体异构体和对映体?
- 保守的结合位点水分子在结合自由能中起着重要作用。
- 如何叠合同类配体序列?
7 如何选取模拟协议
炼金术自由能计算可以大致划分为两大类:“绝对”和“相对“。两者区别在于是否计算单个分子的性质(绝对)还是比较不同但通常相关的分子间的性质(相对)。
7.1 绝对与相对自由能计算的不同之处
7.1.1 相对自由能计算的独特选择
7.1.1.1 拓扑构造
在相对自由能计算中,首先关键的一步是选择何种拓扑构造方法:选用双拓扑、单拓扑还是混合拓扑构造方法。图3中的左边分支展示了单拓扑方法,该方法允许改变原子类型,因此需要引入少量的虚原子。作为对比,图3中的右边分支展示了双拓扑方法,该方法不允许改变原子类型。而混合拓扑方法很少被用到。
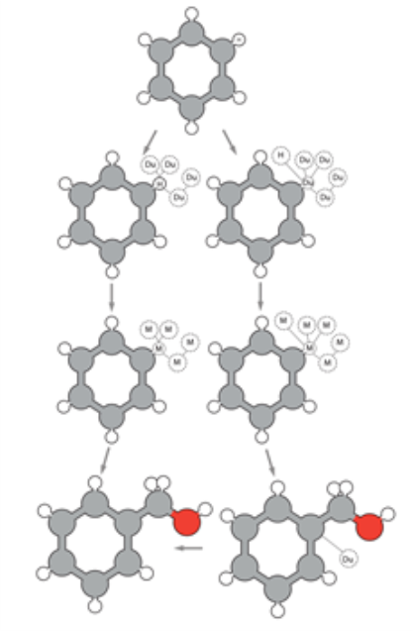
图3. 在炼金术自由能计算中,两种常见的拓扑构造方法
7.1.1.2 原子匹配
在选定了某种拓扑构造方法之后,关键的一步是如何选取不被微扰的相似原子。严格地讲,这一过程实质上包括对相关分子的最大相似子结构(MCSS)的搜索,以找到相似子结构——尽管MCSS搜索的参数将因是否选用了单拓扑还是双拓扑而有所不同。
MCSS方法虽然相对标准,但只考虑拓扑相似性。结合模式的改变实际上可能需要不同的匹配选择,因此在某些情况下,根据原子在三维空间中的位置,可能需要对匹配进行不同的规划。
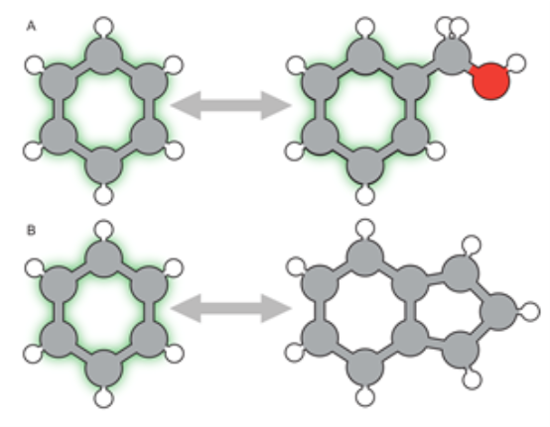
图4. 最大相似子结构(MCSS)匹配:A)绿色标注了 MCSS 区域,这里使用了严格的 MCSS 匹配;B)允许化学键的断裂。
7.1.1.3 化学键的形成与断裂
涉及到环的断裂和形成的相对自由能计算,尤其具有挑战性和问题性。因为相对自由能计算依赖于环中的虚原子的自由能贡献,该贡献不会由热力学循环的不同阶段而抵消。一些方法试图解决这个问题,但一个普适的解决方案尚未被主流使用。
7.1.1.4 微扰路径
基于作为输入的一系列配体分子结构,微扰路径或者网络可以被设计出来。
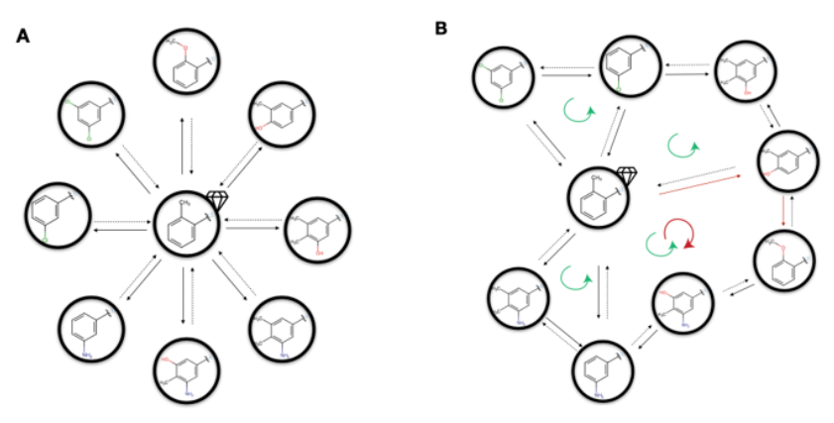
图5. 微扰路径网络例子. A)以晶体结构为中心的星状网络;B)循环闭合型网络;箭头表示微扰的方向;绿色循环圈表示闭合的循环环路;红色循环圈表示较差的循环环路;红色箭头线表示收敛性差的模拟,该模拟导致了引起循环环路不闭合;钻石符号表示选用的晶体结构。
7.1.1.5 约束限制与相对自由能计算
通常,使用SHAKE或LINCS等算法将含氢原子的化学键限制在固定长度,从而允许使用更长的时间步长。但是,典型的分子动力学引擎并不能识别原子类型的变化,或者至少不能正确地包含由改变约束或者约束长度引起的自由能贡献,所以这种相对自由能计算的结果通常是错误的。目前,这个问题最普遍的解决方法是避免在任何涉及约束键的相对自由能计算中使用约束(如果需要,也要使用较小的时间步长,通常为 1fs 左右)。
7.1.2 绝对自由能计算必须处理标准态以及引入约束
7.1.2.1 在绝对自由能计算中的标准态处理
为了解决实际的采样问题和标准态问题,在绝对结合自由能计算中需要引入约束,以保证配体分子在与系统的相互作用被消除时,处于一个定义较好的体积内。
7.1.2.2 几种可能的加约束方法
在实践中,常见的约束类型有很多种。例如,配体和蛋白质之间的简谐距离约束;以及与其工作原理类似但仅在配体离开特定区域时施加力的平底类型约束。还有,由Boresch提出的一组约束也被普遍采用,该方法选取配体相对于受体取向的六个刚体自由度施加约束。甚至还有对整个配体采取RMSD的约束方法。
7.4 绝对与相对自由能计算都需要处理的相似方面
7.4.1 净电荷的改变具有挑战性
目前,还没有工作对电荷修正与其他方法(例如,同时消失离子方法)的仔细比较。因此,在炼金术自由能计算的背景下,正确处理电荷变化的问题仍然是一个值得研究的问题。
7.4.2 考虑多种结合模式
与MD或者自由能计算模拟的时间尺度相比,结合模式转换的时间尺度通常较慢。这意味着从不同可能的结合模式开始的模拟可能会产生不同的结合自由能计算结果。
7.4.3 选取何种采样方案
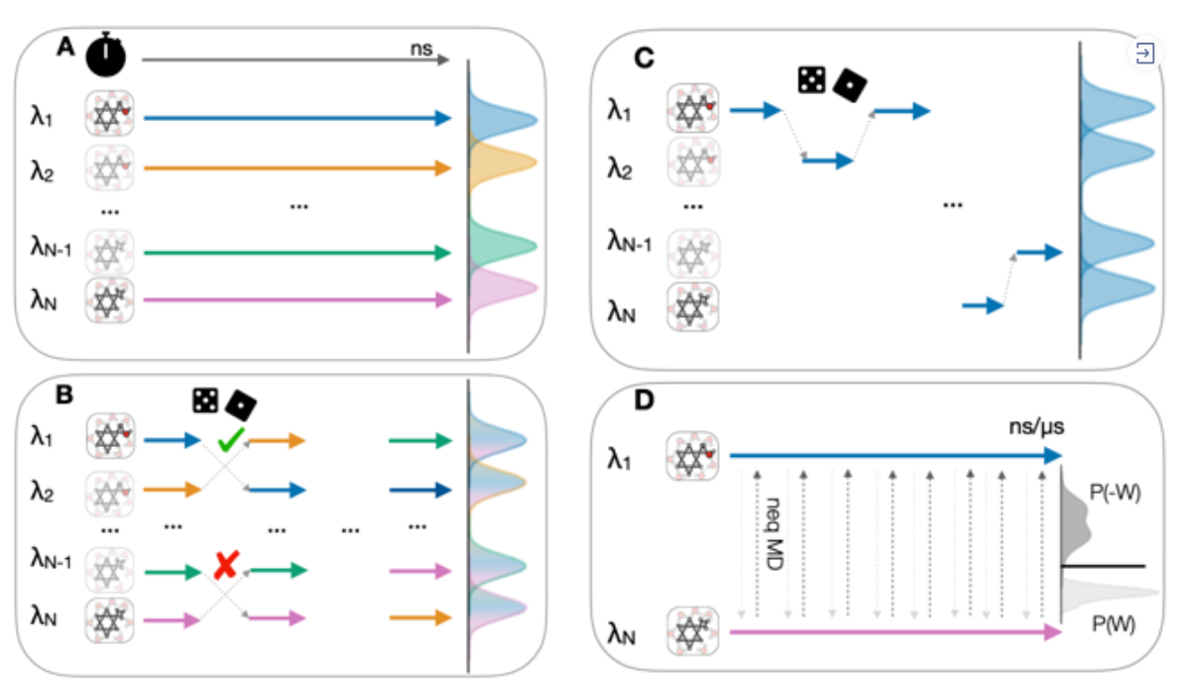
图6. 在不同的软件包中,四种常见的模拟采样方案. A)不同热力学态窗口的多副本并行模拟方案;B)哈密顿副本交换方案;C)对于所有热力学态采样的单副本方案;D)非平衡采样方案。
8 数据分析
8.1 去除采样点的相关性
大多数自由能计算分析方法要求:用于计算自由能差及其统计不确定性的平衡采样结构点之间不应该有相关性。为了实现这一点,模拟保存的结构间隔通常近似地等于或大于统计无效值:
$$
g \equiv 1+2\tau _{eq}
$$
在这里,
$$
\tau _{eq} \equiv \sum_{t = 1}^{T-1}(1- \frac{t}{T}C_t )
$$
其中:
$$
C_t \equiv \frac{\left \langle a_{n}a_{n+t}\right \rangle – \left \langle a{_n} \right \rangle ^2}{\left \langle a_{n}^2 \right \rangle – \left \langle a_{n} \right \rangle ^2}
$$
8.2 自由能计算方法
- 自由能微扰法:Free Energy Perturbation (FEP)
- 热力学积分法:Thermodynamic Integration (TI)
- Bennett接受比例法:Bennett Acceptance Ratio (BAR)
- 多态Bennett接受比例法:Multistate Bennett Acceptance Ratio (MBAR)
关于这几种自由能计算方法,有几条建议:
- 当所有能量差都可以得到时,推荐使用MBAR方法。可以证明,在多态模拟时,该方法可以给出最小误差估计;
- BAR方法是MBAR方法的一种特例。在λ中间态重叠性足够好时,BAR可以给出与MBAR一样好的结果;
- 在中间态窗口足够多的时候,TI方法通常可以给出与MBAR方法一致的结果。但是其积分误差很难预先估计;
- WHAM方法是MBAR方法的近似,没有令人信服的理由证明选用WHAM方法而不用会更好的MBAR方法。
8.3 数据展示的最佳实践
无论你以何种形式展示数据,错误估计都应该包含在你的预测中。建议:同一个自由能计算至少重复三次,以确保数据的可靠性,部分原因是不确定性估计的解析公式通常低估了真实的统计不确定性。
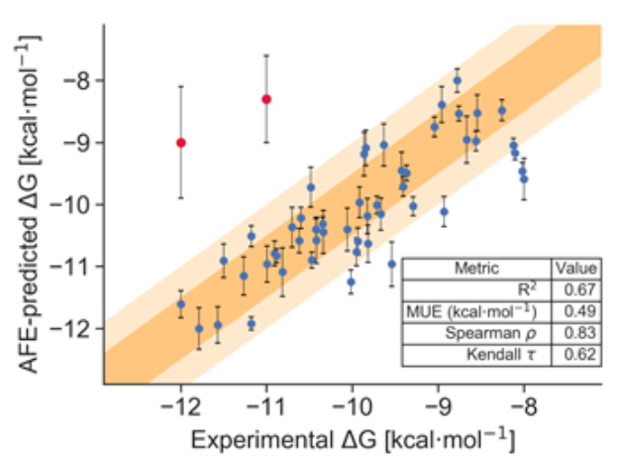
图7. 绘制炼金术自由能预测的推荐实践示例。 该图以kcal/mol为单位显示了预测的和实验的吉布斯自由能之间的关系,其中标准误差表示为误差线(error bar)。 深色和浅橙色区域描绘了1和2 kcal/mol置信区间。 报告了数据的统计指标,通过自举分析确定了95%的置信区间。
在药物研发项目中,该种类型的数据展示可以较好地反映出炼金术自由能预测计算的可靠性,并且可以给出该预测的可信度且作为下一步合成设计的启发。
9 文献
- https://github.com/michellab/alchemical-best-practices
- Mey, A. S. J. S.; Allen, B.; Macdonald, H. E. B.; Chodera, J. D.; Kuhn, M.; Michel, J.; Mobley, D. L.; Naden, L. N.; Prasad, S.; Rizzi, A.; Scheen, J.; Shirts, M. R.; Tresadern, G.; Xu, H. Best Practices for Alchemical Free Energy Calculations. 2020, No. Dlm, 1–48. URL:http://arxiv.org/abs/2008.03067. arXiv:2008.03067
- Chodera, J. D.; Mobley, D. L.; Shirts, M. R.; Dixon, R. W.; Branson, K.; Pande, V. S. Alchemical Free Energy Methods for Drug Discovery: Progress and Challenges. Curr. Opin. Struct. Biol. 2011, 21 (2), 150–160. https://doi.org/10.1016/j.sbi.2011.01.011.
- http://www.ks.uiuc.edu/Training/Workshop/Urbana_2010A/lectures/TCBG-2010.pdf
- Alchemistry wiki: http://www.alchemistry.org/wiki/Best_Practices


