
摘要:准确的力场对于可靠地预测小分子的热力学性质至关重要。然而,AMBER GAFF/GAFF2和OpenFF之类的通用可迁移力场因为扭转参数数量相对有限,在分子动力学模拟和FEP计算的时候可能无法正确表示某些较新的结构或参数化程度较低分子的构象分布。本文介绍了Flare V6如何以全自动的方式生成自定义力场来解决这个问题。
作者:Venkat Ramaswamy/16 August 2022
编译:肖高铿
前言
准确的力场对于可靠地预测小分子的热力学性质至关重要。然而,像 AMBER GAFF/GAFF2 和OpenFF之类的通用可迁移力场因为扭转参数数量相对有限,在分子动力学模拟和FEP计算的时候可能无法正确表示某些较新的结构或参数化程度较低分子的构象分布。计算化学家可以用Flare™以全自动的方式生成自定义力场来解决这个问题。
Flare V6的自定义参数化工作流比以前的版本更加强大。在以前的版本中,用户必须手动选择感兴趣的扭转角并编辑分子以去除不重要的原子以减少与相关的计算成本,这个过程需要用户具有良好的化学知识。在Flare V6中,默认情况下对所有的扭转角进行参数化,分子会在每个感兴趣的扭转角周围自动分割,而不会影响键周边的化学性质。
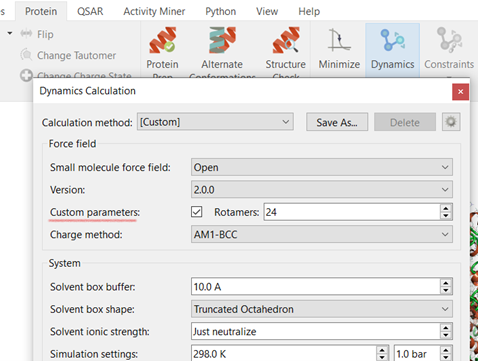
图 1. 只需勾选 Flare Dynamics Calculation 设置中“自定义参数”旁边的框,即可激活 OpenFF 力场的自定义力场生成。 要扫描的旋转异构体的数量也可以根据需要进行更改。
Flare V6自定义参数生成背后的底层组件如下所述。
分子分割
如果分子特别柔性且难以优化,则对分子中的特定二面角进行旋转扫描会变得复杂且耗时。为了解决这个问题,我们根据Wiberg键级 (Wiberg bond Orders ,WBO) 在中心可旋转键周围分割分子。WBO的变化指示了感兴趣键周围的化学变化,母体分子(即配体)被分割成最小的可能片段,直到感兴趣的键的WBO落在特定阈值内。如果不能识别出这样的片段,则将母体分子作为该特定键的片段。这个过程使用我们的OpenFF分子分割器(openff-fragmenter)来实现[1],它使用AmberTools sqm来进行WBO计算。
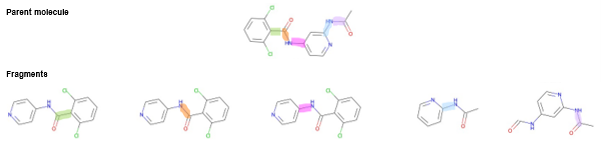
图 2. TYK2抑制剂母体分子及其片段生成示例。母体和片段中相应的扭转角以相同的颜色突出显示。
两面角扫描
传统上,自定义扭转势是用量子力学 (QM) 旋转扫描生成,但这会带来巨大的计算成本,具体取决于分子的柔性(可旋转键的数量)。然而,我们利用了ANI-2X[2],一种机器学习的QM近似,经过专门改进以更好地预测扭转曲线。有了这个,我们以很小的计算成本获得密度泛函理论 (ωB97X/6-31G(d)) 水平的准确性。
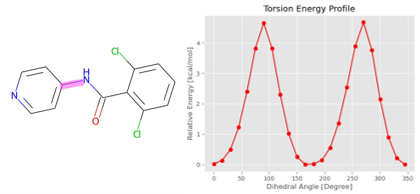
图 3. 使用ANI-2X为片段(左)生成扭转能量分布(右),其中感兴趣的键以洋红色突出显示。整个过程仅需不到4分钟的时间即可完成。
参数优化
最后,将ANI生成的扭转势能用作参考数据以优化扭转参数并使用 ForceBalance[3-4]获得分子的自定义力场。
结果表明,由此生成的自定义力场显著地提高了结合自由能计算的准确性(图 4)。我们工作流的另一个优势是结果的缓存,因此该计算只需对同系列中的公共母核执行一次,从而节省大量的时间和资源用于FEP和分子动力学模拟计算。此外,由于最终的自定义参数存储在本地,你可以与研究相同分子的同事共享,并在任何后续实验中重复使用。
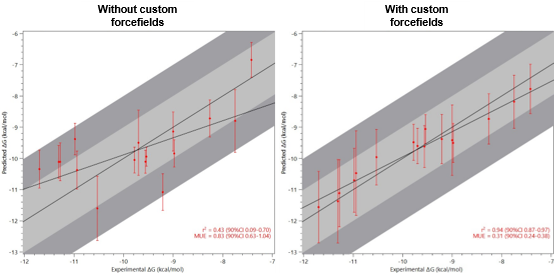
图 4. 示例显示了使用自定义OpenFF力场(右)比标准GAFF2力场(左)对TYK2抑制剂同系物结合自由能计算的准确性提高。结合自由能实验值和预测值的相关性系数 (R2) 从 0.4 提高到几乎为1,而平均无符号误差(MUE)从0.83降低到仅0.31kcal/mol。
预告
下一个版本的Flare将比当前版本显著地提高速度,并在自定义力场工作流中提供一些用户选项。例如,分割任务中基于AmberTools sqm的 WBO计算已被速度更快的半经验程序xTB替代,与当前版本相比,速度至少提高了4-12倍。
另一个改进是进行两面角扫描的方式。在当前版本中,我们使用ANI和geomeTRIC[5]对不同的扭转角分别优化预生成的构象异构体的几何形状。虽然这使得优化任务高度可并行化并且快速,但最终的两面角势能曲线可能看起来并不平滑,并且低能量构象并不总是最优的。因此,我们已将其替换为我们自己的TorsionDrive[6]实现用波前传播获得更准确和更平滑的轮廓。此外,用户现在可以选择他们希望进行扭转扫描的理论水平:机器学习的ANI-2X势能或半经验xTB方法(速度快了不止30 倍)。
参考文献
- Stern, Chaya D., et al. ‘Capturing non-local through-bond effects when fragmenting molecules for quantum chemical torsion scans.’ bioRxiv, doi: 10.1101/2020.08.27.270934
- Devereux, Christian, et al. “Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens.” Journal of Chemical Theory and Computation 16.7 (2020): 4192-4202
- Wang, Lee-Ping, Jiahao Chen, and Troy Van Voorhis. ‘Systematic parametrization of polarizable force fields from quantum chemistry data.’ Journal of chemical theory and computation 9.1 (2013): 452-460
- Wang, Lee-Ping, Todd J. Martinez, and Vijay S. Pande. ‘Building force fields: An automatic, systematic, and reproducible approach.’ The journal of physical chemistry letters 5.11 (2014): 1885-1891
- Wang, L.-P.; Song, C.C. (2016) ‘Geometry optimization made simple with translation and rotation coordinates’, J. Chem, Phys. 144, 214108
- Qiu, Yudong, et al. ‘Driving torsion scans with wavefront propagation.’ The Journal of Chemical Physics 152.24 (2020): 244116


