
摘要:用于苗头化合物与先导化合物的预测性3D定量构效关系(QSAR)方法在早期药物发现阶段非常有价值。在本案例研究中,我们展示了计算机辅助药物设计(CADD)平台Flare1的3D-QSAR在构建磷酸二酯酶10A(PDE10A)抑制剂活性预测模型方面的预测能力和优势。利用1162个配体和77个PDE10A晶体结构的数据集,我们证明了Flare的3D配体叠合工具不仅可以可靠地应用于结构多样的数据集,而且基于配体叠合比基于分子对接的叠合的3D-QSAR显示出更稳健的结果,并且可以在更短的时间尺度内实现。该方法成功地证明了基于配体的方法在分析大型、结构多样的数据集方面不仅仅是一种可行的选择。
理解Flare 3D-QSAR在构建PDE10A抑制剂活性预测模型的预测能力与优势
作者:Jessica Plescia, Stuart Firth-Clark
单位:Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK.
编译:肖高铿
前言
分子对接是一种强大的预测工具,也是药物发现中最常见的基于结构的方法之一。凭借可靠的蛋白质结构信息,它可能是一种非常有用的方法,特别是在与强大的基于配体的方法结合使用时(例如,虚拟筛选结合基于配体的3D性质打分)。然而,有许多因素需要考虑,包括蛋白质构象和柔性2。此外,良好建模的靶标蛋白的晶体结构并不总有,因此需要可靠的基于配体的方法。幸运的是,在这种情况下,我们还有基于配体的方法可用,如基于配体的虚拟筛选和定性或定量结构-活性关系(QSAR)。
Flare的3D-QSAR方法需要基于形状和静电对配体结构进行可靠的叠合。一旦完成配体的构象搜索,Flare就可以使用Cresset场点来识别正、负静电,以及范德华表面(形状)和疏水性来叠合配体。使用这些场点,软件从蛋白靶点的视角(通过静电和形状)来创建配体的模型模式,而不需要蛋白结构3。图1显示了用于治疗精神分裂症的PDE10A抑制剂Mardepodect的场和场点4。

图1. PDE10A抑制剂Mardepodect的生物活性构象(PDB 3HR1)3。左:显示Cresset场。红色=正静电势,蓝色=负静电势。右:显示Cresset场点。红色=正静电势,蓝色=负静电势,黄色=范德华形状,金色=疏水性。
在本案例研究中,Tosstorff等人5的数据集用于展示Flare的3D QSAR方法的预测能力。该数据集由PDE10A的77个晶体结构及其相关的共结晶抑制剂组成,另外还有一组包含1162个具有活性数据的抑制剂,所有抑制剂都通过相同的生化分析进行测试。虽然作者用多模板分子对接研究了这些抑制剂,但本研究将比较Flare的3D-QSAR方法和Flare模板对接(template docking)的结果。
方法
数据准备
在对配体活性数据集进行任何的分析之前,需要先对数据进行准备以用于Flare QSAR实验。为此,使用下述的工作流进行数据准备:
- 从PDB上下载蛋白,并进行蛋白与配体的准备
- 进行序列比对与晶体结构的叠合
- 按子结构对共晶的配体进行分类
- 将共晶配体按子结构分开,并将抑制剂叠合到其上
第一个任务是准备77个晶体结构,(1)将共晶的参比配体以生物活性构想方式提取出来,(2)准备蛋白结构以作为后续实验的排除体积。为此,从支持信息中复制PDB编码,并将相应的结构下载到Flare中。完成导入蛋白结构之后,很明显存在“过量”的蛋白单体,为了简单起见可以删除。这可使用Flare Python Cookbook中的一个代码片段完成。蛋白质表单中只保留有A链,然后开始准备蛋白结构,包括确定活性位点和提取参比配体。然后将来自支持信息的活性数据与配体表单中的分子合并。
接下来,对PDE10A晶体进行序列比对和叠合。在Flare中的蛋白叠合也会导致相应的共晶配体也被叠合。不出所料,这些蛋白的序列一致性均大于98%,最大RMSD为0.55Å。图2显示了晶体结构5SDU以及经RMSD评估最为多样的八种晶体结构。从叠合中可以清楚地看出,它们在3D空间中仍然非常靠近。
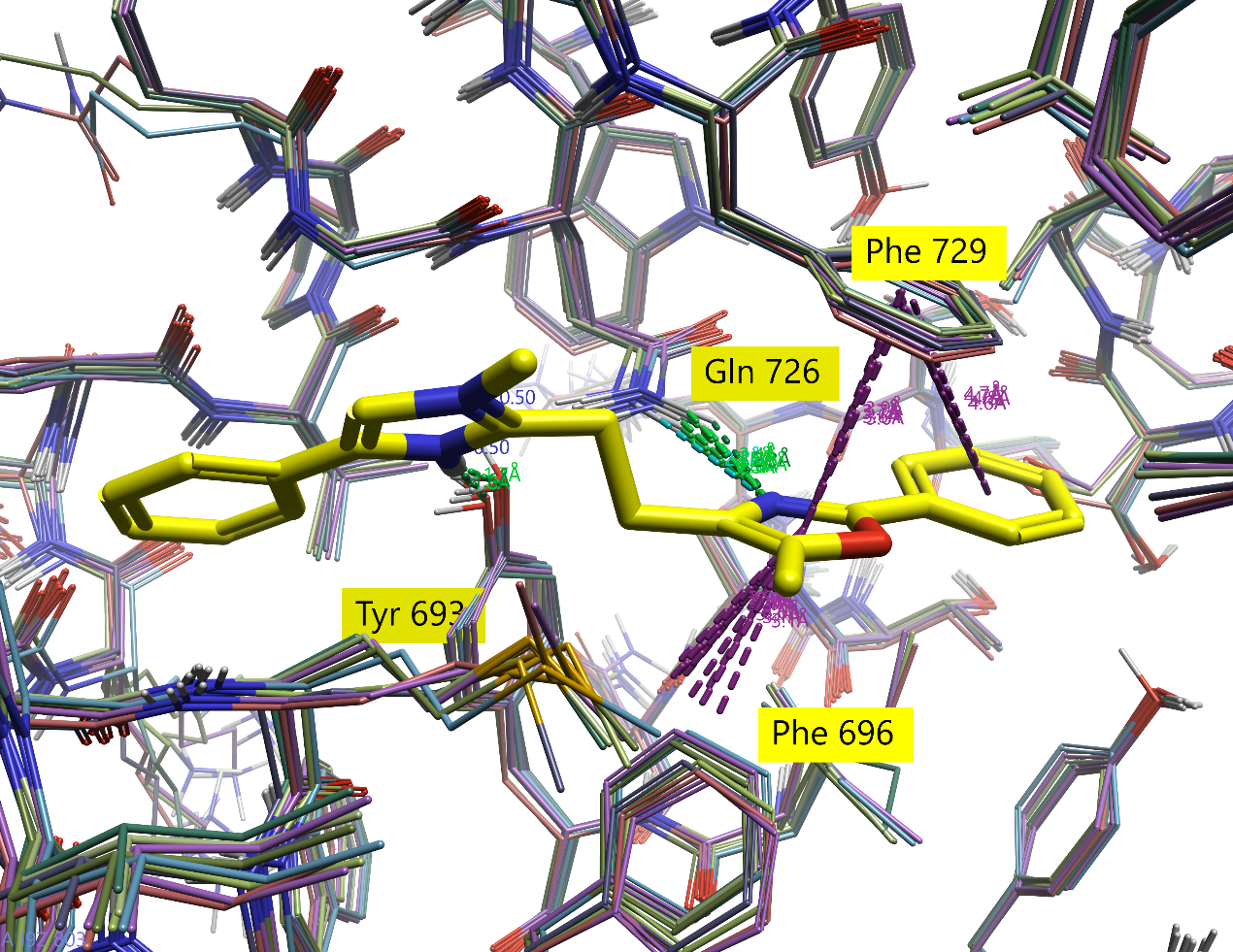
图2. 5SDU及其参比配体(黄色)通过RMSD叠合到八种最多样的晶体结构上:5SDY, 5SE1, 5SEF, 5SEW, 5SEW, 5SF2, 5SFB, 5SFS。
如图2所示,参比配体与每个晶体结构产生了关键、稳定的相互作用,即与配体发生稳定相互作用的关键残基的位置在所有晶体结构中是保守的。
接下来,将具有PDE10A活性的1162个配体导入到Flare中并转化为2D结构。作者先前将它们分为三类子结构,分别标记为氨杂芳基_c1_酰胺(aminohetaryl_c1_amide)、芳基_cl_酰胺_c2_杂芳基(aryl_c1_amide_c2_hetaryl,)和c1_杂芳基_烷基_c2_杂芳基(c1_hetaryl_alkyl_c2_hetaryl)。每个类别的代表性配体如图3所示。

图3. 每一类活性最强的配体及其Cresset场点(其上为2D结构)。左: aminohetaryl_c1_amide;中: aryl_c1_amide_c2_hetaryl;右: c1_hetaryl_alkyl_c2_hetaryl。
基于配体的技术:最大公共子结构配体叠合用于QSAR研究
为了进行QSAR实验,每个子集都需要与相应的参比分子进行叠合。选择最佳的参考分子是配体叠合工作流中非常重要的一步;有必要选择代表整个数据集的参比分子,以便优化最终叠合并因此优化QSAR模型的准确性。例如,选择完全未取代的参比分子,例如具有未取代苯环的配体,将导致邻位和间位取代的配体在两个方向上的叠合,从而在最终的QSAR模型中产生更多的噪音。在这种情况下,尽管每个数据集中的大多数配体都有非常相似的骨架,但R基团变化很大。因此,为每个子集选择了9个参比分子,以最好地代表配体结构的多样性。图4说明了这个选择过程。3D视图显示了为aminohetaryl_c1_amide子集选择的九个参比分子中的四个。

图4. aminohetaryl_c1_amide子集的4个参比分子
这四种晶体结构在分子的西向具有不同取代的吡唑。这种选择为形状和静电提供了几种不同的参比场点模式,使得在aminohetaryl_c1_amide子集中不同取代的配体与参比分子中最紧密匹配的那个进行叠合。相反,如果选择A IF5 803唯一的参比分子(图4,左下),这可能会导致在叠合过程中配体绕着酰胺-吡唑键发生不必要的旋转。重复这一过程,为三个配体子集选择出具有代表性的参比分子。
一旦为每个子集选择了9个具有代表性的参比配体,就可进行构象搜寻和分子叠合。由于数据集的大小和一些配体的柔性,构象搜寻选择了“Accurate but Slow”设置,以增加构象总数并降低能量窗口截止值。然后使用“MCS”和“Permissive”(杂交匹配)规则进行分子叠合:MCS叠合首先发现每个参比分子和每个配体之间的共性。MCS区域被冻结,分子的其余部分进行构象搜索,然后使用形状和静电叠合到参比分子上。在本案例研究中,由于可以获得蛋白的结构信息,因此将蛋白作为为排除体积,即对占据活性位点的叠合施加惩罚。为排除体积选择的PDB是与最强活性的参比配体共结晶的那个。图5显示了三个配体子集的叠合结果。
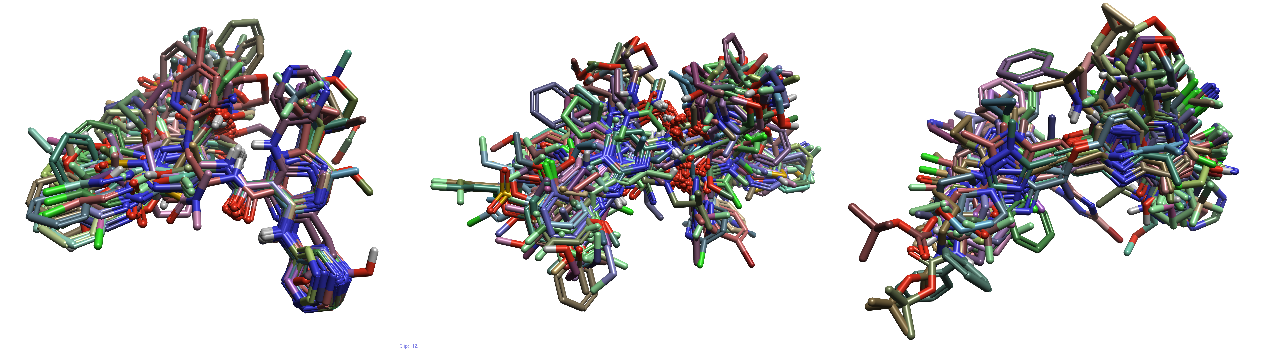
图5. 三个配体子集的叠合。左: aminohetaryl_c1_amide, 中: aryl_c1_amide_c2_hetaryl, 右:c1_hetaryl_alkyl_c2_hetaryl
从图5中可以清楚地看出,虽然每个配体集中的2D子结构都有一些共同点,但它们仍然相当多样化。这些叠合用于每个配体子集的QSAR模型搭建。
基于配体的技术:基于静电场与形状的配体叠合用于QSAR研究
在Flare中可用的另一种叠合类型是“标准”(Normal)叠合,使用Cresset的基于场的叠合和打分方法,仅基于分子静电和形状而不是化学结构进行叠合。这种类型的叠合更具生物学相关性,因为它忽略了2D结构,只关注3D形状和静电。它对不同的配体结构也更方便,当我们将含有1159个配体的数据集作为一个整体时可看到这一点。
为了进行这种筛选,使用精确但缓慢(Accurate but Slow)模式的构象搜索,来源于每个结构子集活性最强的三个参比化合物作为Flare标准叠合的参比分子。图6显示了1159个配体的总体配体排列。

图6. 1159个配体与每个结构子集的三个活性最强参比分子的标准模式叠合。左:3D结构的叠合。右:打开场点。关键官能团(左)和场点聚集区域(右)用圆圈突出显示。
尽管1159个配体的叠合看起来有些无序,但有几个关键官能团非常紧密叠合的区域,例如吡啶/嘧啶氮,仲酰胺氮,酰胺羰基和羟基/醚。当将场点时打开显示时,这些叠合在视觉上变得更加突出,并且可以看到正、负场的聚集。
逐个配体的观察可以更好地可视化这些叠合。图7显示了来自每个子结构集合的一个配体叠加到活性位点中的九个参比分子中的三个,每个子结构组一个。
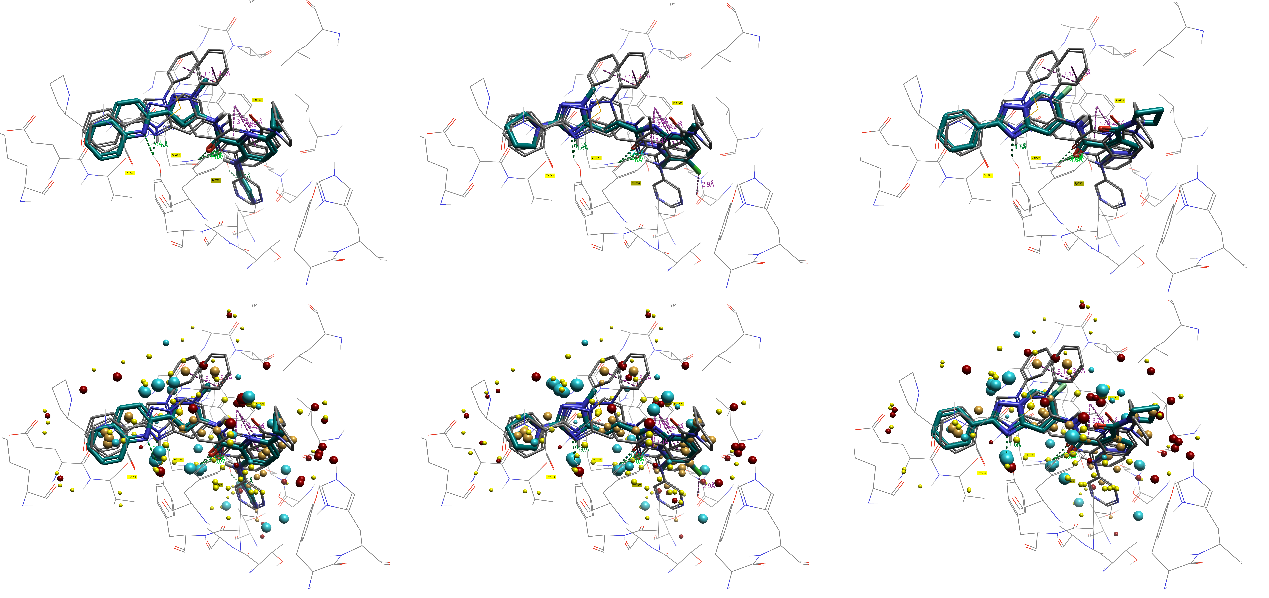
图7. 5SE7活性位点以及叠合到三个参比分子(灰色)上的配体(茶绿色):左:aminohetaryl_c1_amide, 配体46, pIC50 = 10.1;中:c1_hetaryl_alkyl_c2_hetaryl, 配体300, pIC50 = 9.48;右: aryl_c1_amide_c2_hetaryl, 配体204, pIC50 = 9.11
从图7可以清楚地看出,尽管2D结构存在差异,但叠合还是成功的。不仅2D结构相对对齐,而且场点聚集也很明显。此外,保留了与活性位点残基的关键相互作用:所有三个配体都与Tyr693和Gln726形成氢键,并与Phe729形成芳香相互作用,就像共晶的参比配体一样。
基于结构的技术:分子对接配体叠合用于QSAR研究
由于Tosstorff等人[5]的原始研究使用多模板对接的结合模式来进行QSAR预测,因此使用Flare的单模板选项进行分子对接来重现该QSAR研究。对于每个配体子集,使用活性最强的相应参比配体作为模板对接的种子来引导对接结果。还对先前提到的具有最高RMSD偏差的九个晶体结构进行了系综对接,以允许蛋白结构的最大变化。图8显示了每个配体子集的对接结构的叠合。不出所料,对接结合模式的变化明显高于基于配体的叠合,因为配体在指定的对接区域内有更大的自由度来与蛋白互补(6.0Å活性位点能量网格由所选的参比配体周围的6Å半径来定义)。

图8. 各个配体子集叠合好的对接结合模式。左:aminohetaryl_c1_amide,中:aryl_c1_amide_c2_hetaryl,右:c1_hetaryl_alkyl_c2_hetaryl
QSAR回归模型的构建
在建立QSAR模型之前,将每个子集的10%(或“标准”配体叠合的完整数据集的20%)隔离出来用做验证集,剩余的配体被划分为训练集和测试集,比例为70/30,活性范围分布均匀。用于MCS配体叠合和对接叠合的训练集/测试集/验证集的划分如下:aminohetaryl_c1_amide子集为285/122/45,aryl_c1_amide_c2_hetaryl子集为264/113/42,c1_hetaryl_alkyl_c2_hetaryl子集为181/78/29。由于“标准”配体叠合没有将配体按结构子集划分,因此将20%配体移出用于验证集,其余的则分为训练集和测试集。最终划分为649/278/232,作为训练集/测试集/验证集。由于SMILES含有两个单独的配体作为外消旋混合物,因此原始1162个中3个配体被删除掉。
Flare的自动回归选项用于构建所有可能的回归模型,即支持向量机(SVM),随机森林(RF),高斯过程(GP),k近邻(kNN),多层感知器(MLP),并选择结果最佳的模型。还探索了共识模型(Consensus model),该模型计算了五个单独回归模型的预测平均值。Flare中可用的另一个模型是Field QSAR,这是一种回归方法,它使用Cresset的形状和静电3D描述符,不仅可以建立预测性模型,还可以进行3D可视化以阐释空间体积和正/负静电势对活性的贡献。本研究还探索了Field QSAR实验。表1显示了QSAR结果,包括给出最佳预测性能的模型和由此产生的相关性,以及验证集的相关性和均方根误差(RMSE)。Field QSAR使用留一法交叉验证方法,但不输出单独的训练集交叉验证相关性。
结果
基于MCS叠合的QSAR
第一轮QSAR模型构建聚焦在MCS法叠合的配体上,结果如表1所示。
表1. 每个配体子集用MCS法叠合后的QSAR模型

从表1可以清楚地看出,尽管配体结构叠合的变化很大,但预测模型的表现相当好。虽然c1_hetaryl_alkyl_c2_hetaryl子集的活性预测值与实验值的相关性并不理想,但就数据集大小而言,表征预测值与实验值之间差异的RMSE相对于所有配体子集而言相当低。平均而言,通过回归模型或Field QSAR计算的预测值与实验值之间的差值小于1 pIC50值(RMSE平均值=0.83)。对于我们基于配体的活性预测方法来说,这个数据非常有前景,并且在随机、跨子结构标签、临时分割数据集地评估所有配体时,优于Tosstorff等人5基于结构的方法(RMSE=1.04-3.31)。
为了查看结果是否可以改进,检查了aminohetaryl_c1_amide数据集,并通过旋转单键针对参比配体单独调整了叠合结果。该模型的预测结果的R2仅增加了0.02,即3%。对于最小的数据集c1_hetaryl_alkyl_c2_hetaryl配体,重复了这种调整,但预测结果R2再次增加了0.02,即5%。尽管在数据子集内的结构变化存在较大的视觉差异,但这种微不足道的改进突显了人们对Flare叠合的信心。
基于静电场与形状叠合的QSAR
标准模式配体叠合的QSAR建模结果如表2和图9所示。
表2. 基于静电场与形状叠合的QSAR模型
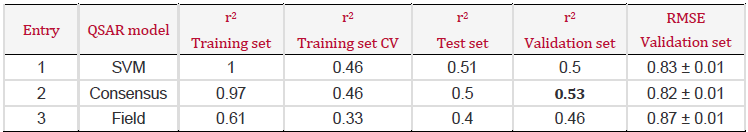
如本文结构多样性数据集所示,QSAR数据进一步支持配体叠合提供更加稳健的QSAR模型的观点。此外,所有1159个化合物的“标准”叠合结果与每个子结构子集的MCS叠合结果相当,证明了Cresset 3D形状和静电描述符的价值。“标准”模式和MCS模式叠合的平均RMSE值是可比的:平均RMSE值分别为0.83和0.84。对于这个特定的数据集,当对新的预测集进行MCS模式或“标准”模式叠合时,可以在1 pIC50值内可靠地预测新配体的活性。

图9. 用标准模式叠合的验证集的预测值与实验值比较。左: r2= 0.50 Support Vector Machine;中:r2= 0.53 Consensus;右: r2= 0.46 Field QSAR.
基于分子对接叠合的QSAR
基于分子对接叠合QSAR模型的建立类似于基于配体的实验,结果如表3所示。
表3. 基于对接叠合的QSAR模型

考虑到对接配体的叠合比基于配体的叠合要无序得多,对接叠合方法测试集的相关性较低也就不足为奇了。然而,在aryl_c1_amide_c2_hetaryl子集上,基于结构和基于配体的配体准备之间的具有最大差异,对接方法的RMSE=1.13,考虑到3D结构叠合的巨大差异,这仍然令人印象深刻。人们已经证实,通常,使用基于配体叠合方法而不是通过分子对接叠合方法准备的配体数据集的3D QSAR模型可以产生更可靠的QSAR模型。本文的这些QSAR实验结果支持了这一结论:与由对接配体构建的模型相比,由基于配体的叠合构建的3D QSAR模型在预测抗PDE10A活性方面更可靠。此外,鉴于实验的复杂性,基于结构的对接实验比基于配体的叠合计算需要的时间要长得多。
结论
活性预测是药物发现中备受追捧的工具,并且存在许多基于配体和基于结构的方法可供选择。当蛋白质结构可用时,许多研究小组专注于基于结构的方法,例如自由能微扰(FEP),分子动力学(MD)或分子对接。当蛋白质结构不可获得时,必须使用基于配体的方法,例如药效团建模或QSAR。然而,在本文的案例中,对配体信息和结构生物学信息的同时利用可导致协同QSAR结果;共晶配体作为参比分子可用于指导配体叠合,从而产生高度可靠的QSAR模型。
可靠的QSAR模型需要稳健可靠的配体数据集,最好来自同一研究小组或机构,并使用相同的生化分析进行测试。幸运的是,公开的数据集5符合这些标准,并允许进行几个QSAR实验。使用Cresset基于配体的叠合方法以及配体静电和形状3D描述符,所得预测模型可以在1 pIC50的误差内准确地预测配体活性,证明了该方法的稳健性。
尽管没有一种正确的方法来分析数据集并生成模型来预测配体活性,但这本的研究表明,QSAR对于这组PDE10A配体来说一直是可靠的。
参考文献
- Flare™, version 7, Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130
- Meng, X.-Y., et al. Molecular Docking: A powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011, 7(2), 146–15.7. doi.org/10.2174/157340911795677602
- Cheeseright, T., et al. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J Chem Inf Model 2006, 46, 2, 665–676. doi.org/10.1021/ci050357s
- Verhoest, P. R., Chapin, D. S., Corman, M. et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J Med Chem 2009, 52, 16, 5188–96. doi.org/10.1021/jm900521k
- Tosstorff, A., Rudolph, M. G., et al. A high quality, industrial data set for binding affinity prediction: performance comparison in different early drug discovery scenarios. J Comp-Aided Molec Des 2022, 36, 753–765.
获取试用、商务合作,请联系我们
立即申请免费的Flare试用版,用Flare 3D-QSAR来分析您自己的项目!此外,我们还提供FTE技术服务,由我们的专家帮您完成计算任务。


