
摘要:文章讨论了二甲基膦酰基衍生物在药物化学中的细胞膜渗透性。膦酰基作为电子吸引基团,因其相对不亲脂的特性而受到关注。勃林格殷格翰的研究表明,二甲基膦酰基可能影响药物的渗透性,因为它是优秀的氢键受体,这一点通过pKBHX值得到证实。尽管如此,许多含膦酰基的药物通过形成分子内氢键来提高渗透性。文章还探讨了“游离型”膦酰基化合物的渗透性,并指出在增加分子量的同时提高亲水性对保持良好的渗透性有一定限度。通过几个案例分析,文章得出结论,膦酰基化合物在保持其他性质不变的情况下,仍可获得良好的通透性,但在大而不太亲脂性的分子中添加膦酰基会导致通透性下降。最后,文章提到膦酰基在药物设计中的创造性应用,包括作为氢键受体的稳定性和生物活性构象中的重要性。
作者:Gilles Ouvry. November 25, 2024. NRG Therapeutics. available at: https://www.linkedin.com/pulse/permeability-phosphine-oxides-gilles-ouvry-fyy9e
编译:肖高铿/2024-12-27
在人们发现了ALK抑制剂Brigatinib1之后,在过去10年里,膦氧化物(phosphine oxides)就越来越多地出现在临床候选药物中。作为取代基,二甲基膦酰基在克雷格图(Craig plot,是指一种用于展示药物化学中取代基性质的图表)中无疑是最强的电子吸引基团之一,并且也是相对较不亲脂的官能团之一。

幸运的是,对于药物化学界而言,勃林格殷格翰(BI,Boehringer Ingelheim)的科学家们决定在药物化学背景下对基于磷元素的基团进行全面研究,并将他们的研究成果发表在2020年出版的《Journal of Medicinal Chemistry》杂志上2。我无论如何强调这一成果的重要性都不为过,尤其是他们合成并全面剖析了许多类似药物结构的衍生物。下面是伊马替尼(imatinib)衍生物的logD与Caco-2的实验测量结果。
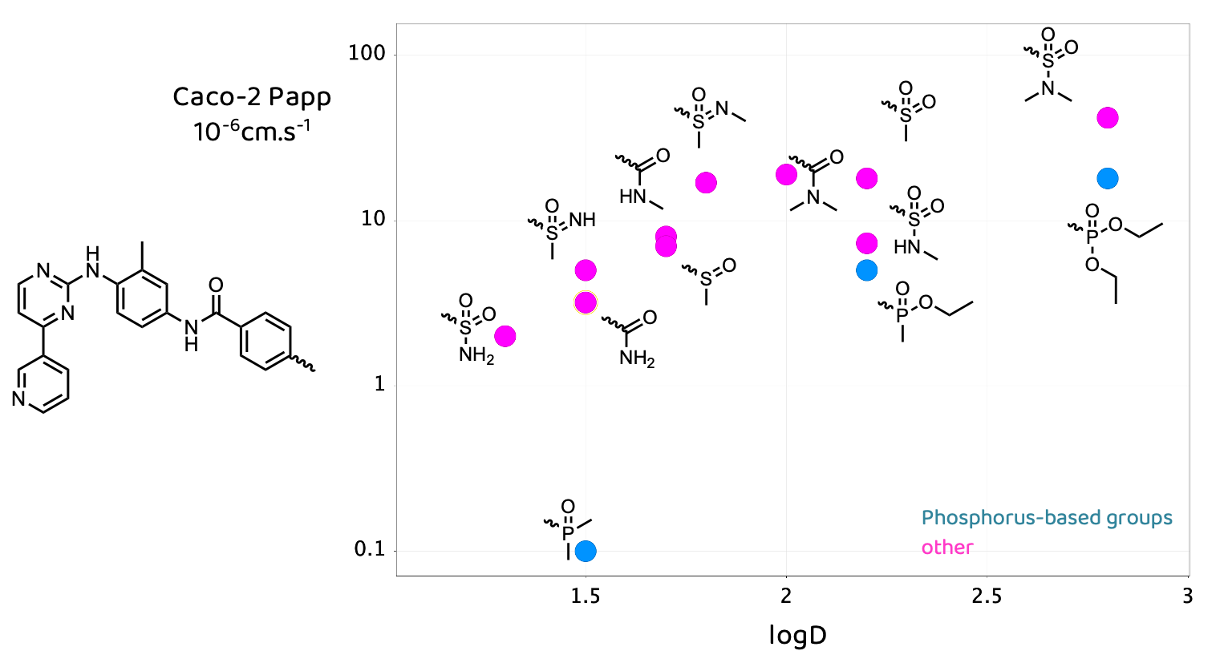
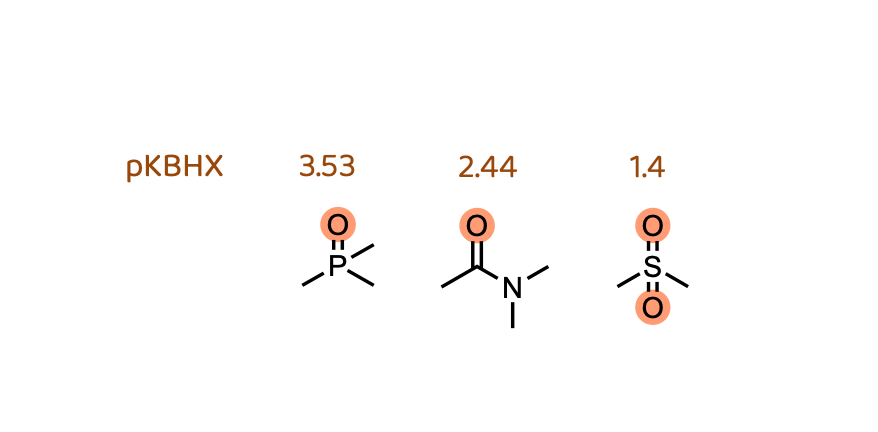
值得注意的是,二甲基膦酰基似乎“破坏”了这种相关性。换句话说,考虑到它的亲脂性,我们本应预期它更具渗透性。一个可能的原因是膦酰基是极其优秀的氢键受体。Desai等人3已被证明这会对膜渗透性带来负面影响。这一性质可以被测量,并且被称为 pKBHX(或 pKβ)。想要了解更多信息/数据,可以查阅Laurence等人4原始的 pKBHX 实验测量汇总,Peter Kenny5 提供了一个更新的汇总可以下载。膦酰基作为氢键受体,比酰胺优秀10倍、比砜类优秀100倍。在将它添加到你的分子上时,你确实需要考虑这一点。
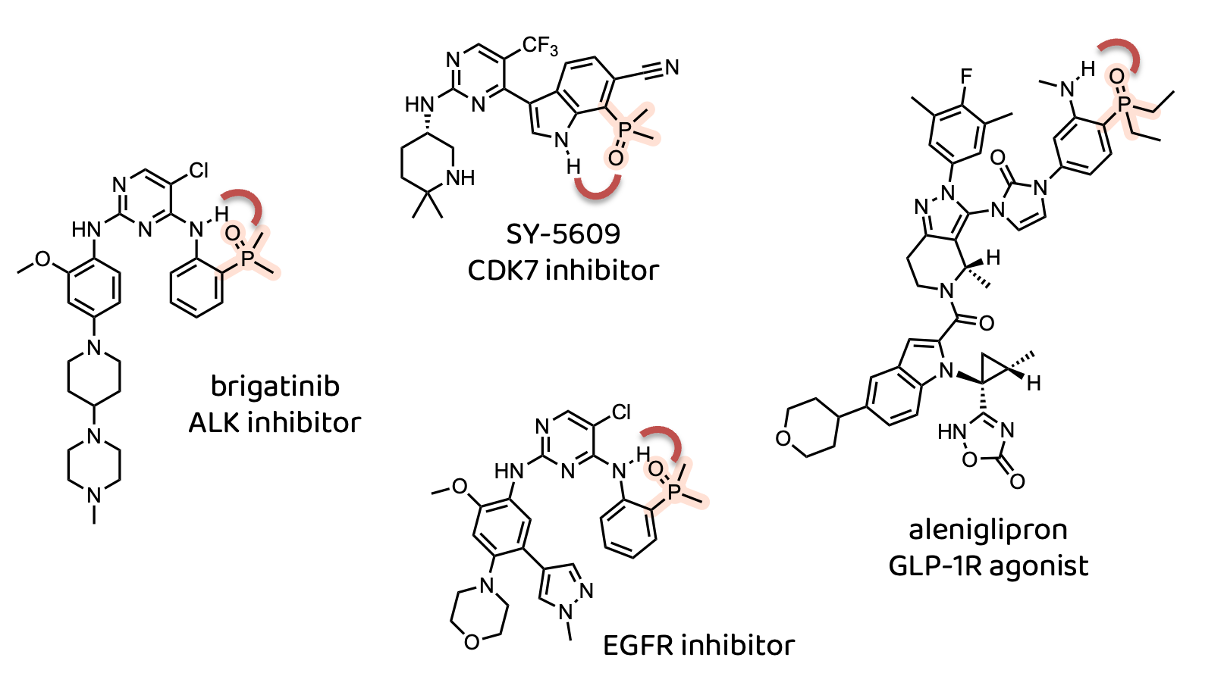
那么我们不禁要问,含膦酰基药物的渗透性会受到多大的影响。有趣的是,大多数携带膦酰基的药物/临床候选药物中的膦酰基都参与了分子内氢键(Intra Molecular Hydrogen Bond,IMHB)的形成,比如Ariad的ALK抑制剂Brigatinib1、Syros的CDK7抑制剂SY-56096、阿斯利康的EGFR抑制剂7以及GLP-1R激动剂aleniglipron。分子内氢键确实会屏蔽膦酰基并提高其渗透性。
这不禁让人质疑,分子内氢键(IMHB)是否是绝对的前提条件?为了尝试回答这个问题,我查阅了相关文献。我应该先说明一下,在药物化学领域虽然有很多关于膦酰基使用的报道,但我并没有找到任何关于渗透性或者疏水参数logD的实验数据。因此,为了解答这个问题,我在文章中寻找在细胞水平比酶学水平活性下降的案例,并将其与其它官能团进行比较——显然这种方法存在一定的局限性,还有多种其他能解释细胞水平活性下降的原因,但这仍然是一个合理的起点。
简要总结一下,好消息是,含有“游离型(即不参与分子内氢键)”膦酰基的化合物可以具有完美、充足的渗透性——但你仍然需要谨慎。正如Mike Waring8在15年前在他的开创性关于渗透性的论文中所指出的,如果你想保持良好的渗透性,在增加分子量的同时提高亲水性是有一定限度的。
第一个例子来自上海药物研究所Sun等人9的PARP抑制剂。在这个案例中,膦酰基衍生物的活性下降明显大于其他类似物(如氧环和醇类),更接近于带电的氮杂环丁烷。正如前面提到的,从通透性的角度来看,强亲水性取代基在如此大的分子(分子量:546)上特别不被容忍,这并不令人意外。
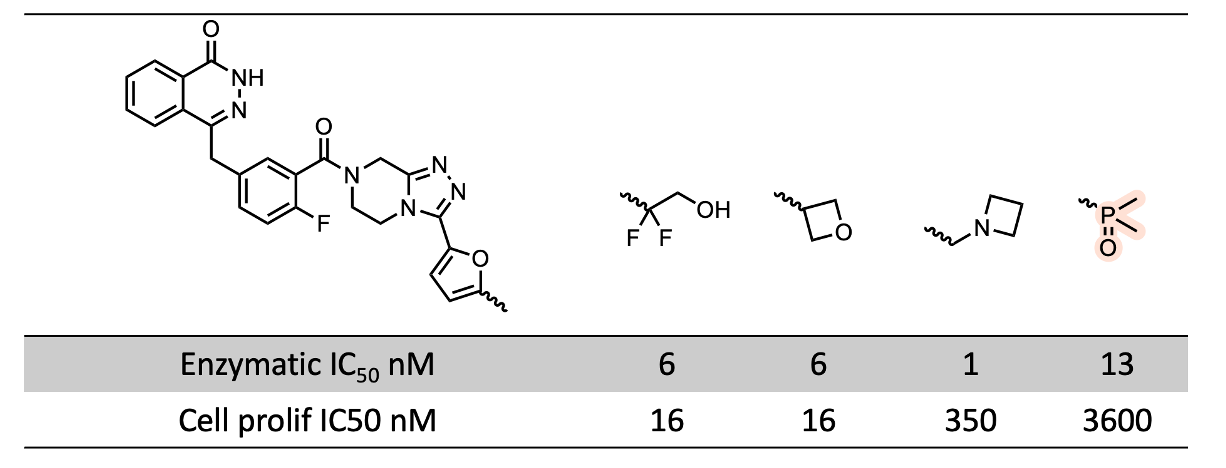
下一个案例来自加思科药业(JacoBio)的SHP2抑制剂项目10,感谢Praful Chovatia提供了这一线索。膦酰基衍生物的活性下降再次显著高于其他类似物,考虑到其分子大小(分子量:530),并且在这种情况下,氢键供体(HBD)的数量进一步加剧了这种现象。
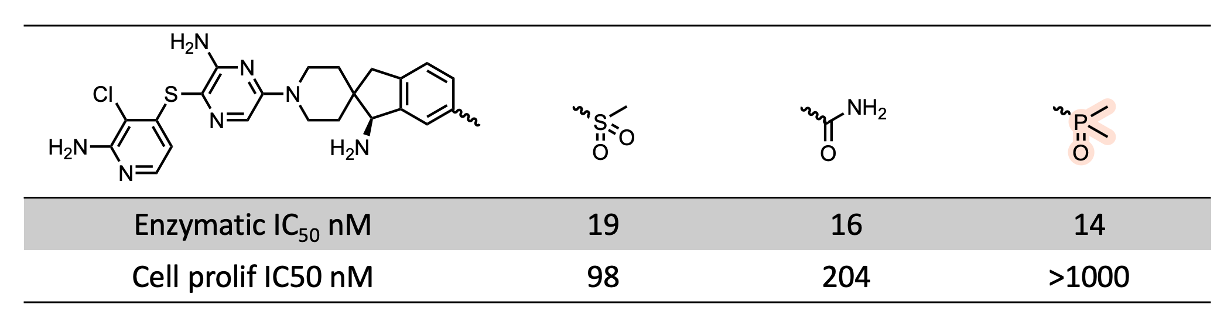
转向较小的和/或更亲水的分子,我们来看看Celgene的JAK2抑制剂11。尽管膦酰基衍生物的活性略高于砜类衍生物,但在这种情况下,活性下降可能更容易管理。
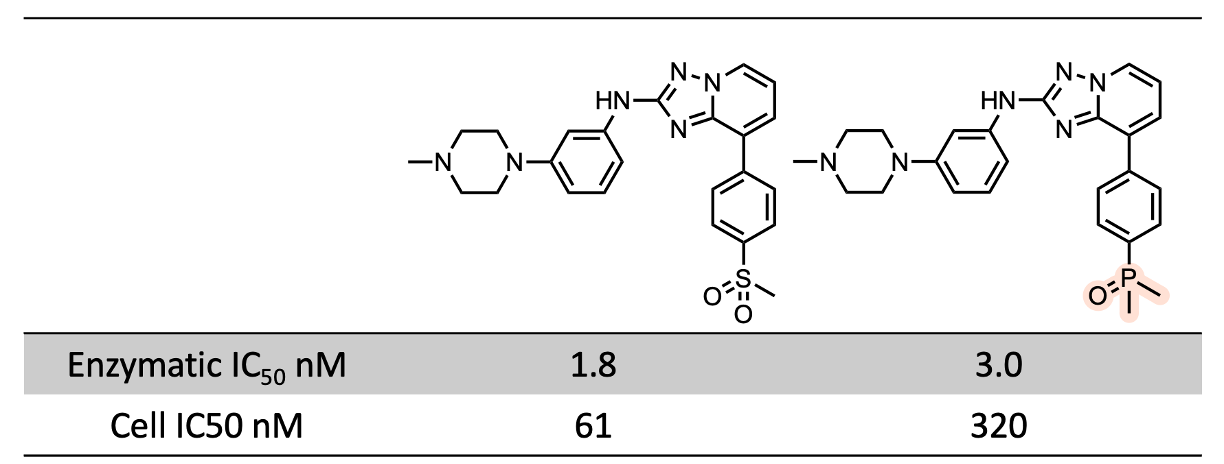
作为较早采用膦酰基的科学家,Ariad的研究人员成功地在多个项目中使用了这类基团,包括以下BCR-Abl的例子12。在这个案例中,膦酰基衍生物与吡啶类似物之间的活性下降没有差异。尽管化合物的分子量很大(590),但这些化合物亲脂性如此的强(膦酰基的XlogP \(>\) 5),以至于能够容的下膦酰基。
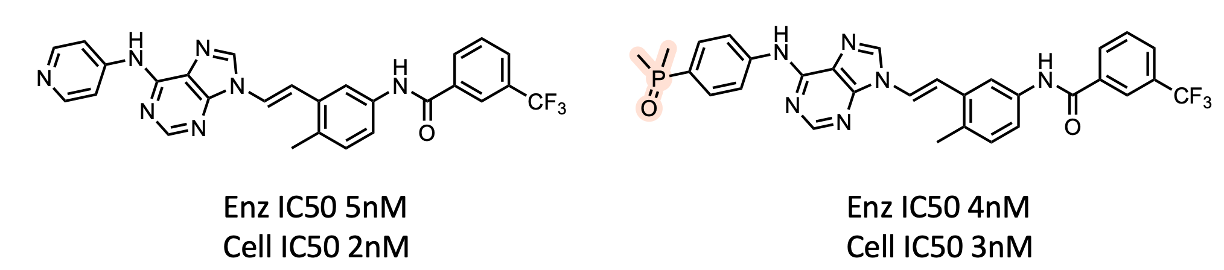
最后两个例子分别来自拜耳的科学家及其ATR抑制剂项目13和密歇根大学的科学家们(又一次!)及其胚胎外胚层发育(embryonic ectoderm development,EED)抑制剂项目14。对于这两个项目,我能够从发表的文章中绘制出所有化合物。我认为这表明,在通透性下降方面,这些项目的膦酰基衍生物的行为没有明显差异(即绿色星标不是异常值)。这些分子较小(分子量\(<\)450),只有一个氢键供体(HBD),这可能使它们处于理想的通透性空间中。
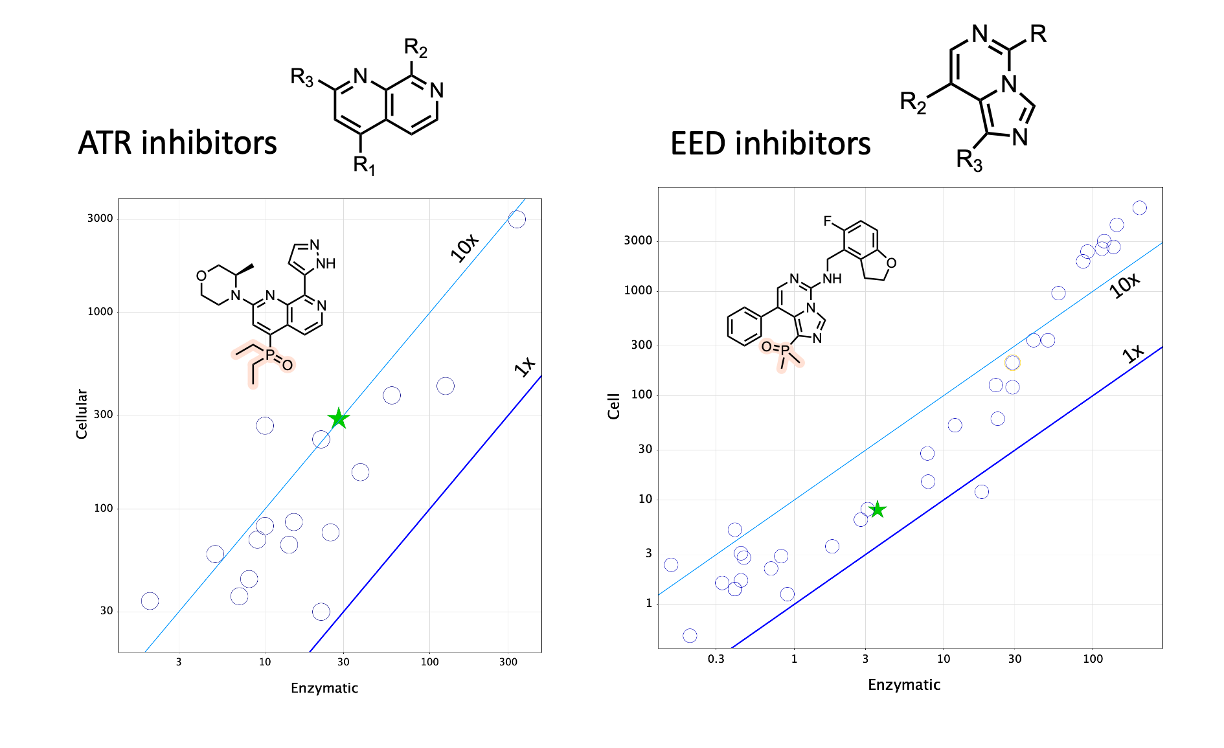
我发现最后两个例子相当令人安心。是的,膦酰基化合物确实极性很强且是非常好的HBA(氢键受体),但在保持其他性质不变的情况下,你仍然可以得到良好的通透性。然而,将它们添加到大而不太亲脂性的分子中时会导致可预测的通透性下降。令人欣慰的是,你可以通过精心设计的分子内氢键来缓解这种情况….
但等等,还有更多…
膦酰基(以及其他含磷原子基团)真的能激发药物化学家的创造力。勃林格殷格翰(BI)的科学家们利用膦酰基的高度极性/低通透性的性质发现了BI 1265162——一种吸入型ENaC抑制剂,要求具有较低的通透性15,感谢Pierre Sierocki提供了这个例子。罗氏的科学家们巧妙地在TLR7抑制剂(WO2024/013205)中使用了膦酰基16。例如,一个2-磺酰胺嘧啶类似物在化学上是不稳定的,但一个膦酰基衍生物则是稳定的….这绝对值得记住。最后,虽然不是有意设计的,膦酰基的强大HBA能力对AZ的BFl-1抑制剂的生物活性构象中9元环分子内氢键至关重要17。

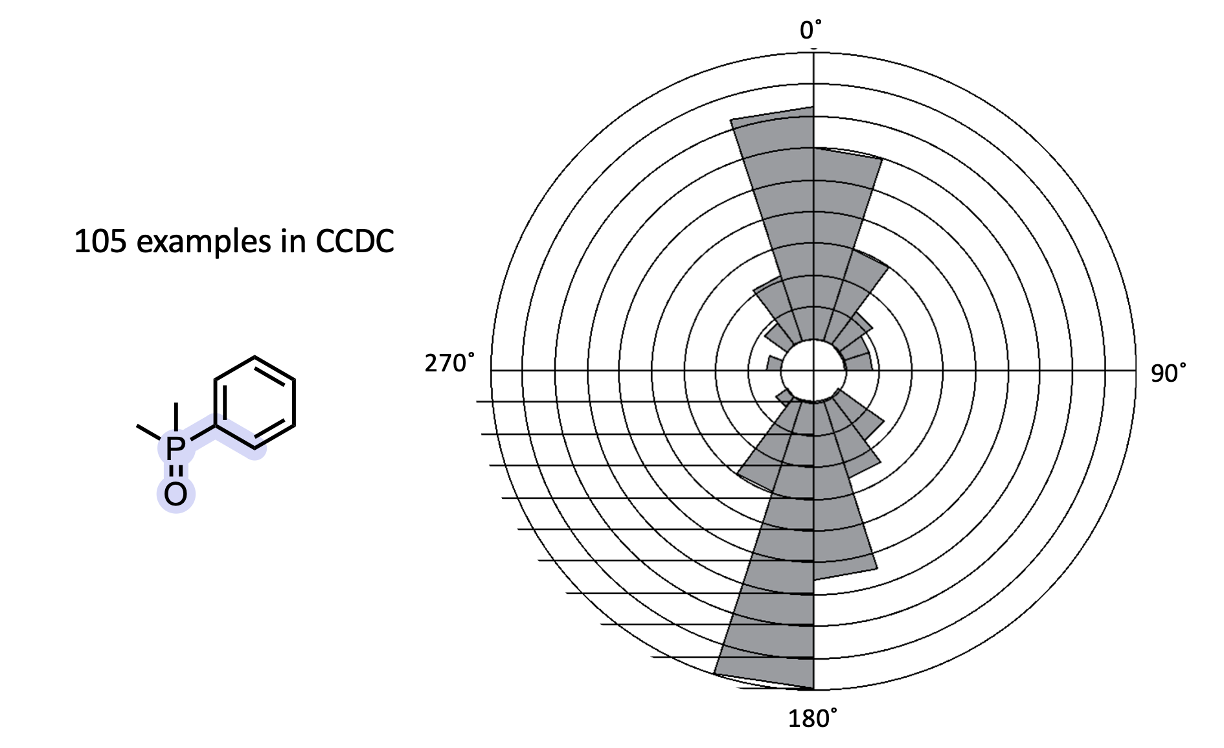
如果没有一个小小的CCDC扭转角分析图,这将不是一个完整的LinkedIn帖子… 我只是想用一个从小分子X-衍射世界发现的膦酰基扭转角分析图来结束这段分享。与酰胺不同,芳环被酰胺取代时,它们往往会失去共平面性(三级酰胺几乎从不共面);而膦酰基似乎更倾向于保持共平面…尽管我的猜测是这更多是因为空间位阻而非电子效应的原因。
文献
- Huang, W.-S. et al. (2016) “Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase,” Journal of Medicinal Chemistry, 59(10), pp. 4948–4964. Available at: https://doi.org/10.1021/acs.jmedchem.6b00306.
- Finkbeiner, P., Hehn, J.P. and Gnamm, C. (2020) “Phosphine Oxides from a Medicinal Chemist’s Perspective: Physicochemical and in Vitro Parameters Relevant for Drug Discovery,” Journal of Medicinal Chemistry, 63(13), pp. 7081–7107. Available at: https://doi.org/10.1021/acs.jmedchem.0c00407.
- Desai, P. v., Raub, T.J. and Blanco, M.-J. (2012) “How hydrogen bonds impact P-glycoprotein transport and permeability,” Bioorganic & Medicinal Chemistry Letters, 22(21), pp. 6540–6548. Available at: https://doi.org/10.1016/j.bmcl.2012.08.059.
- Laurence, C. et al. (2009) “The p K BHX Database: Toward a Better Understanding of Hydrogen-Bond Basicity for Medicinal Chemists,” Journal of Medicinal Chemistry, 52(14), pp. 4073–4086. Available at: https://doi.org/10.1021/jm801331y.
- Kenny, Peter W (2020). Hydrogen bond basicity data: text file with measured pKBHX values, isomeric SMILES strings and literature references. figshare. Dataset. https://doi.org/10.6084/m9.figshare.12084183.v1
- Marineau, J.J. et al. (2022) “Discovery of SY-5609: A Selective, Noncovalent Inhibitor of CDK7,” Journal of Medicinal Chemistry, 65(2), pp. 1458–1480. Available at: https://doi.org/10.1021/acs.jmedchem.1c01171.
- Finlay, M.R. v. et al. (2021) “Potent and Selective Inhibitors of the Epidermal Growth Factor Receptor to Overcome C797S-Mediated Resistance,” Journal of Medicinal Chemistry, 64(18), pp. 13704–13718. Available at: https://doi.org/10.1021/acs.jmedchem.1c01055.
- Waring, M.J. (2009) “Defining optimum lipophilicity and molecular weight ranges for drug candidates—Molecular weight dependent lower logD limits based on permeability,” Bioorganic & Medicinal Chemistry Letters, 19(10), pp. 2844–2851. Available at: https://doi.org/10.1016/j.bmcl.2009.03.109.
- Sun, Y. et al. (2023) “YCH1899, a Highly Effective Phthalazin-1(2 H )-one Derivative That Overcomes Resistance to Prior PARP Inhibitors,” Journal of Medicinal Chemistry, 66(17), pp. 12284–12303. Available at: https://doi.org/10.1021/acs.jmedchem.3c00821.
- Ma, C. et al. (2024) “Discovery of JAB-3312, a Potent SHP2 Allosteric Inhibitor for Cancer Treatment,” Journal of Medicinal Chemistry, 67(16), pp. 13534–13549. Available at: https://doi.org/10.1021/acs.jmedchem.4c00360.
- Dugan, B.J. et al. (2012) “A Selective, Orally Bioavailable 1,2,4-Triazolo[1,5- a ]pyridine-Based Inhibitor of Janus Kinase 2 for Use in Anticancer Therapy: Discovery of CEP-33779,” Journal of Medicinal Chemistry, 55(11), pp. 5243–5254. Available at: https://doi.org/10.1021/jm300248q.
- Huang, W.-S. et al. (2009) “9-(Arenethenyl)purines as Dual Src/Abl Kinase Inhibitors Targeting the Inactive Conformation: Design, Synthesis, and Biological Evaluation,” Journal of Medicinal Chemistry, 52(15), pp. 4743–4756. Available at: https://doi.org/10.1021/jm900166t.
- Lücking, U. et al. (2020) “Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 with Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models,” Journal of Medicinal Chemistry, 63(13), pp. 7293–7325. Available at: https://doi.org/10.1021/acs.jmedchem.0c00369.
- Rej, R.K. et al. (2020) “EEDi-5285: An Exceptionally Potent, Efficacious, and Orally Active Small-Molecule Inhibitor of Embryonic Ectoderm Development,” Journal of Medicinal Chemistry, 63(13), pp. 7252–7267. Available at: https://doi.org/10.1021/acs.jmedchem.0c00479.
- Betzemeier, B. et al. (2024) “Discovery and development of BI 1265162, an ENaC inhibitor for the treatment of cystic fibrosis,” European Journal of Medicinal Chemistry, 265, p. 116038. Available at: https://doi.org/10.1016/j.ejmech.2023.116038.
- Sabnis, R.W. (2024) “Novel Phosphorylpurinone Compounds for Treating Cancer,” ACS Medicinal Chemistry Letters, 15(4), pp. 441–442. Available at: https://doi.org/10.1021/acsmedchemlett.4c00087.
- Lucas, S.C.C. et al. (2024) “Identification and Evaluation of Reversible Covalent Binders to Cys55 of Bfl-1 from a DNA-Encoded Chemical Library Screen,” ACS Medicinal Chemistry Letters, 15(6), pp. 791–797. Available at: https://doi.org/10.1021/acsmedchemlett.4c00113.
原创文章,作者:小墨,如若转载,请注明出处:《二甲基膦酰基衍生物的细胞膜渗透性》http://blog.molcalx.com.cn/2024/12/27/permeability-of-phosphine-oxides.html


