
摘要:磷光材料是OLED领域开发的重点之一,通过对有机染料的磷光发射波长计算,可以预测化合物作为磷光材料的潜质。已经发现Ir(ppy)3为一绿色磷光材料,在甲苯中发射波长为513nM。本教程以Ir(ppy)3为例,用Gaussian软件预测其发射波长,并与实验观察到的波长进行比较以评估计算的精度。
肖高铿/2016-05-22
一. 磷光发射的过程
磷光材料是OLED领域开发的重点之一,已经发现Ir(ppy)3 (CAS94928-86-6, 图2)为一绿色荧光材料,在二氯甲烷与四氢呋喃中发射波长为509nM,在甲苯中发射波长为513nM(王晓峰等,2015)。通过对有机染料的磷光发射波长计算,可以预测化合物作为磷光材料的潜质(Unger, Y., et al. 2013)。为了说明计算过程,本教程以Ir(ppy)3为例,用Gaussian 09 D.01预测其磷光发射波长,并与实验观察到的波长进行比较以评估计算的精度。
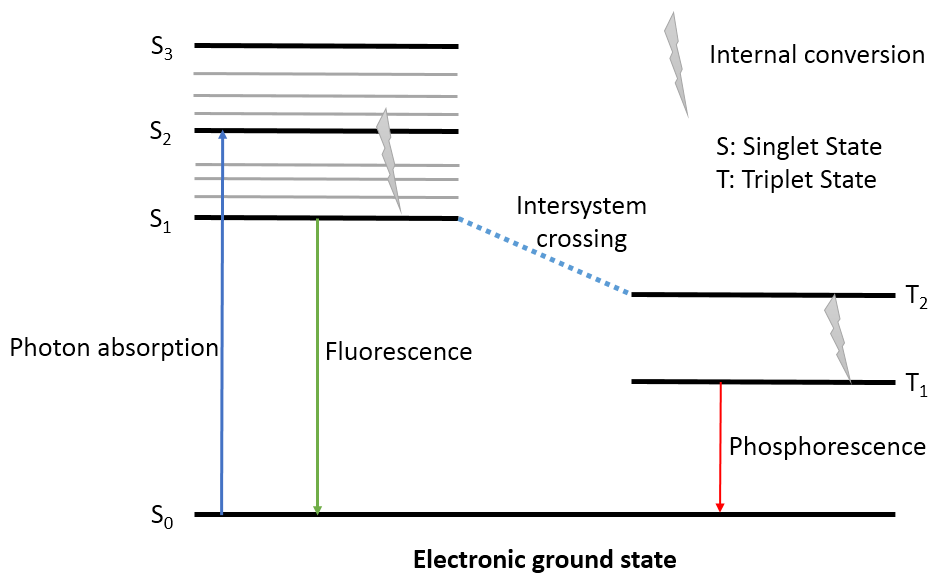
图1. 激发态与荧光、磷光
如图1所示,化合物从激发三重态T1退激为基态单重态S0时以光子的形式释放出能量,假定这份能量全部转化为光子,该光子的波长就对映着发射的磷光波长。在该过程中,T1与S0具有完全一样的构象,先计算T1的构象与能量,再用T1构象计算S0能量,两者能量差值即是波长。
T1为三重态,其能量可以用TD-DFT(td=triplet)或用DFT(设置自旋多重度为3)这两种方法计算获得。在TD-DFT中计算激发态,必然会计算基态S0的能量,因此对结构进行TDDFT的优化,就直接得到激发态与基态的能量差(发射的磷光波长)。而采用后者计算,需要单独再用单点能计算S0以获得能量差。Unger, Y.(2013)等人就采用DFT(自旋多重度=3)法计算多个化合物的磷光发射波长,建立线性方程来预测化合物的磷光,可以将预测误差控制在5nM之内。
计算采用的软件与方法:我们采用Gaussian 09 D.01推荐的标准理论水平APFD/6-311+G(2d,p)进行计算。其中Ir原子采用LANL2DZ基组,计算时用PCM溶剂化模型将甲苯考虑进来。计算流程与Unger, Y.(2013)等人的方法一致。
二,计算步骤
1,结构搭建
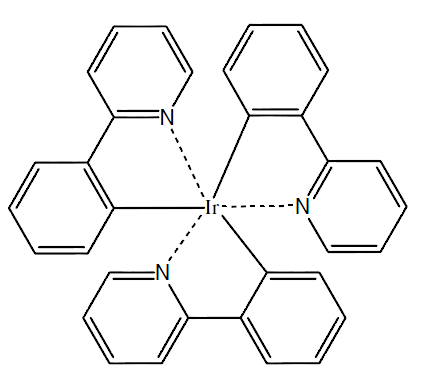
用GaussView5搭建化合物结构,其平面结构如图2所示。
 |
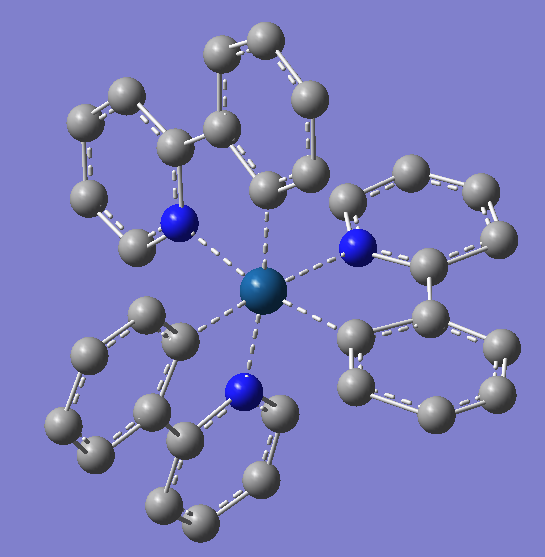 |
图2,Ir(ppy)3的2D结构与3D结构(氢原子未被展示)
当然,也可以从晶体结构数据库下载这个结构进行计算。
2,T1的结构优化与能量计算
将搭建的结构进行进行激发三重态优化获得T1结构,对应的Gaussian 09作业文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 | %mem=24GB %nprocshared=12 %chk=D:\work\demo\oled\t1.chk # opt apfd/genecp integral(ultrafinegrid) scrf=(iefpcm,solvent=toluene) Structure: Ir(PPY)3; T1 structure Optimization 0 3 C 12.24000000 1.41800000 1.91000000 C 13.01300000 1.88900000 2.92700000 H 12.84600000 2.69700000 3.35600000 C 14.41500000 -0.09200000 2.72800000 H 15.15800000 -0.56900000 3.02100000 C 13.72200000 -0.61300000 1.74100000 H 13.95900000 -1.42100000 1.34400000 C 12.59800000 0.12400000 1.31800000 C 11.76600000 -0.36500000 0.23800000 C 12.03400000 -1.60600000 -0.48600000 H 12.75900000 -2.16400000 -0.31800000 C 11.08400000 -1.84300000 -1.44300000 H 11.22100000 -2.58900000 -1.98000000 C 9.99200000 -1.13800000 -1.69600000 H 9.37500000 -1.40500000 -2.33700000 C 9.81200000 0.02400000 -0.94900000 H 9.06400000 0.55300000 -1.10000000 C 10.50700000 3.84700000 2.37500000 C 11.09100000 5.12300000 2.22700000 H 11.61200000 5.29200000 1.47500000 C 10.91700000 6.12000000 3.14600000 H 11.30300000 6.95300000 2.99300000 C 10.19100000 5.91900000 4.28300000 H 10.08100000 6.60400000 4.90500000 C 9.62200000 4.68400000 4.49600000 H 9.15300000 4.52100000 5.28100000 C 9.74500000 3.66500000 3.53200000 C 9.09000000 2.34600000 3.70000000 C 8.23400000 1.95000000 4.70000000 H 8.00600000 2.54700000 5.37500000 C 7.71300000 0.69100000 4.70800000 H 7.10100000 0.45400000 5.36600000 C 8.07500000 -0.22600000 3.76800000 H 7.77700000 -1.10700000 3.81400000 C 8.90100000 0.19700000 2.74900000 H 9.12100000 -0.41000000 2.08000000 C 11.57100000 3.19600000 -0.35900000 C 12.94000000 3.39900000 -0.50900000 H 13.51600000 3.09900000 0.15700000 C 13.48200000 4.04600000 -1.64300000 H 14.40000000 4.17300000 -1.71900000 C 12.62500000 4.48800000 -2.64100000 H 12.97600000 4.90000000 -3.39600000 C 11.27200000 4.32400000 -2.52800000 H 10.71000000 4.63600000 -3.20100000 C 10.72000000 3.68400000 -1.39000000 C 9.28500000 3.50800000 -1.19900000 C 8.25900000 3.89500000 -2.08900000 H 8.47200000 4.29800000 -2.90000000 C 6.94900000 3.67500000 -1.75500000 H 6.28700000 3.92500000 -2.35600000 C 6.57800000 3.10200000 -0.57100000 H 5.68600000 2.95500000 -0.35000000 C 7.62500000 2.74800000 0.29100000 H 7.41400000 2.36700000 1.11200000 N 10.71300000 0.39700000 -0.00400000 N 8.93000000 2.93300000 -0.01700000 N 9.40600000 1.44800000 2.67200000 Ir 10.59600000 2.24700000 1.14100000 C 14.16800000 0.98400000 3.30300000 H 14.87147199 1.17536613 4.08620251 Ir 0 LANL2DZ **** C H N -O 0 6-311+G(2d,p) **** Ir 0 LANL2DZ radii=bondi !此行特意空白 |
3. S0能量计算
T1->S0释放光子的过程非常快以至于化合物的构象还没来得及变化,S0具有与T1一样的构象,因此用基态单重态方法对T1结构进行单点计算即可。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | %mem=24gb %NprocShared=12 %oldchk=D:\work\demo\oled\t1.chk %chk=D:\work\demo\oled\s0.chk # apfd/genecp geom=check integral(ultrafinegrid) scrf=(iefpcm,solvent=toluene) S0 energy computation 0 1 Ir 0 LANL2DZ **** C H N -O 0 6-311+G(2d,p) **** Ir 0 LANL2DZ radii=bondi !此行特意空白 |
三. 计算结果
| T1能量(au) | S0能量(au) | 能量差(au) | 计算波长(nM) | 实验波长(nM) |
|---|---|---|---|---|
| -1540.2226696 | -1540.3098830 | 0.0872134 | 522.51 | 513 |
文献报道Ir(ppy)3在室温下甲苯溶液中的磷光发射波长为513nM(王晓锋等2015),计算值为522左右,比实验值偏红了~9.5nM。
四. 相关教程
荧光计算教程:http://blog.molcalx.com.cn/2016/08/14/gaussian-fluorescence-tutorial.html
五. 文献
[1]王晓峰,左国防,李志锋,李会学.(2015) 位移谐振子模型对三(2-苯基吡啶)合铱磷光光谱影响的理论研究. 物理化学学报,2015,31(9):1667-1676.
[2]Unger, Y., et al. (2013). “Prediction of the emission wavelengths of metal-organic triplet emitters by quantum chemical calculations.” Journal of Organometallic Chemistry 748: 63-67.
六. 联系我们
电邮:info@molcalx.com
电话:020-38261356
单位:广州市墨灵格信息科技有限公司
网站:http://www.molcalx.com.cn/gaussian


