
摘要:本文介绍了电离势和亲和势的相关概念,并以苯分子为例,详细介绍了如何利用Gaussian程序(高斯)计算分子的电离势和亲和势。
作者:陈宇
日期:2019-01-17
一. 基本概念
1.1 电离势(Ionization Potential)
通常也指第一电离势,是处于气态的中性原子或分子失去一个电子,变成 +1 价离子时的能量变化(也称为电离能),定义为:
IP = E(N-1) – E(N)
其中IP为电离势,E(N-1)为+1价离子基态时的电子能,E(N)为中性原子或分子基态时的电子能,N为体系总电子数。当电离势值为正数时,表明中性分子失去一个电子需要吸收能量。
当+1价离子再失去一个电子,形成+2价离子时,其能量变化也称为第二电离势,定义为:
IP = E(N-2) – E(N-1)
以此类推,可以求得第三,第四…..电离势。
电离势有两种(图1所示):垂直电离势(Vertical Ionization Potential, VIP)和绝热电离势(Adiabatic Ionization Potential, AIP)。垂直电离势是指只优化N电子体系的分子构型,然后使用这个构型计算N和N-1电子体系的能量,再相减求得电离势。而绝热电离势是指分别优化N和N-1电子体系的分子构型并求得能量,然后相减求得电离势。
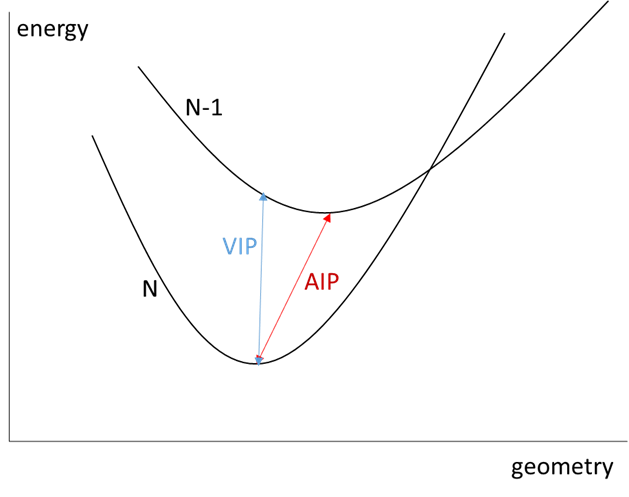
图1. 电离势示意图
1.2 亲和势(Electron Affinity, EA)
是指第一亲和势,是处于气态的中性原子或分子得到一个电子,变成 -1 价离子时的能量变化,定义为:
EA = E(N) – E(N+1)
其中EA为亲和势,E(N+1)为-1价离子基态时的电子能,E(N)为中性原子或分子基态时的电子能,N为体系总电子数。当亲和能值为正数时,表明中性分子获得一个电子时会释放能量。
当-1价离子再得到一个电子,形成-2价离子时,其能量变化也称为第二亲和势,定义为:
EA = E(N+1) – E(N+2)
以此类推,可以求得第三,第四…..亲和势。
亲和势同样有两种(图2所示):垂直亲和势(VEA)和绝热亲和势(AEA)。垂直亲和势是指只优化N电子体系的分子构型,然后使用这个构型计算N和N+1电子体系的能量,再相减求得亲和势。而绝热亲和势是指分别优化N和N+1电子体系的分子构型并求得能量,然后相减求得亲和势。
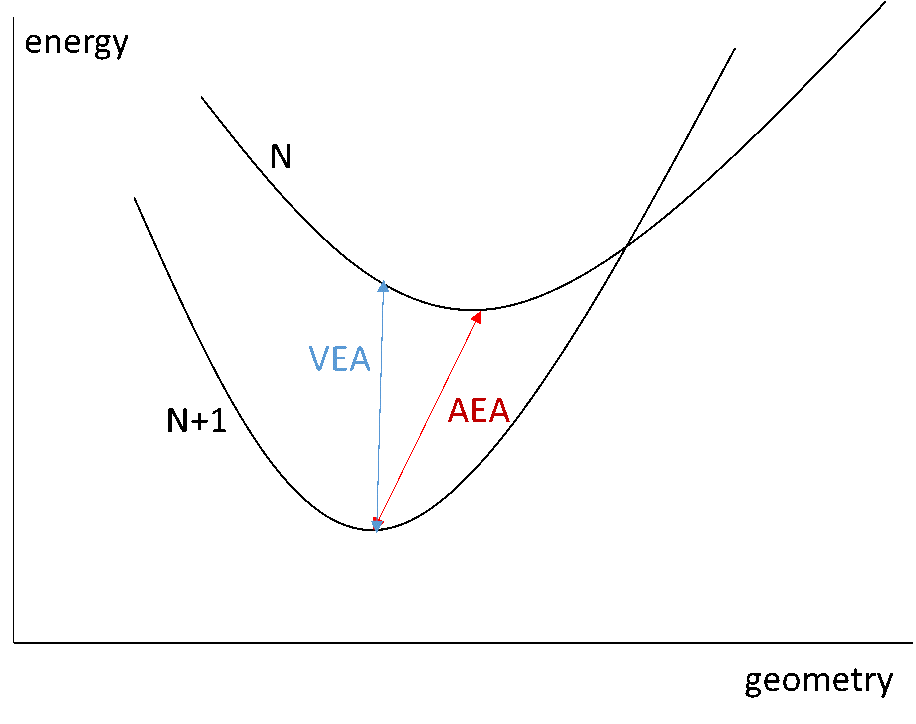
图2 亲和势
需要注意的是:计算电离势和亲和势时都需要用如下公式进行热校正,但对于垂直电离势或垂直亲和势,由于中性分子和离子采用同样的结构,因此热校正值可以认为近似相等,在这种情况下,也可以直接用电子能相减。
ΔEtherm = ΔEelec + Ecor
其中ΔEtherm为热校正后的电子能,ΔEelec为电子能,Ecor为热校正值。
二.计算分子电离势和亲和势
在这里我们以计算第一电离势为例,所以下面的步骤中是优化中性分子,如果计算第二电离势,依照上面的公式即可。
计算垂直电离势和垂直亲和势的步骤:
- 首先优化中性分子的结构。
- 以中性分子结构为基础,用相同的方法基组对+1价离子(为了计算电离能)或-1价离子(为了计算亲和势)进行单点能计算。
- 依照电离势或亲和势公式,能量相减。
计算绝热电离势和绝热亲和势的步骤:
- 优化中性分子和相应的+1价离子(为了计算电离势)或-1价离子(为了计算亲和势)的结构,同时进行频率分析。
- 依照电离势或亲和势公式,能量相减。
在本文中,我们以苯分子作为例子,分别计算垂直电离势(以及亲和势)和绝热电离势(以及亲和势)。
图3. 苯分子的结构
2.1 计算苯分子的垂直电离势和垂直亲和势
2.1.1 第1步:优化中性苯分子结构
优化中性苯分子结构的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | %nprocshared=8 %mem=1GB # apfd/6-311+G(d,p) opt freq Ph-neutral 0 1 C -0.44991508 0.72156196 0.00000000 C 0.94524492 0.72156196 0.00000000 C 1.64278292 1.92931296 0.00000000 C 0.94512892 3.13782196 -0.00119900 C -0.44969608 3.13774396 -0.00167800 C -1.14729708 1.92953796 -0.00068200 H -0.99967408 -0.23075504 0.00045000 H 1.49475292 -0.23095104 0.00131500 H 2.74246292 1.92939296 0.00063400 H 1.49532892 4.08996496 -0.00125800 H -0.99981808 4.09002496 -0.00263100 H -2.24690108 1.92972096 -0.00086200 !上面特意是一行空白行 |
2.1.2第2步:计算失去一个电子和得到一个电子后形成的离子单点能
以第一步优化的结构为基础,分别计算中性苯分子失去一个电子和得到一个电子后形成的离子单点能。
计算中性苯分子失去一个电子后形成的阳离子单点能的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | %nprocshared=8 %mem=1GB # apfd/6-311+G(d,p) Ph-cation-SPE 1 2 C -1.37961400 -0.19041100 0.00000100 C -0.52478300 -1.28978400 0.00002800 C 0.85467500 -1.09938500 -0.00001000 C 1.37960600 0.19046400 0.00000600 C 0.52483300 1.28976400 0.00001200 C -0.85471700 1.09935300 -0.00001900 H -2.45522900 -0.33907200 -0.00000900 H -0.93398400 -2.29559700 -0.00002300 H 1.52111800 -1.95666900 -0.00005300 H 2.45523900 0.33898800 -0.00002300 H 0.93391200 2.29562500 0.00002700 H -1.52105300 1.95672100 -0.00001800 !上面特意是一行空白行 |
因为中性分子失去一个电子后电荷为1、自旋多重度为2,所以第7行的电荷与自旋多重度分别设为1与2。
计算中性苯分子得到一个电子后形成的阴离子单点能的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | %nprocshared=8 %mem=1GB # apfd/6-311+G(d,p) Ph-anion-SPE -1 2 C -1.37961400 -0.19041100 0.00000100 C -0.52478300 -1.28978400 0.00002800 C 0.85467500 -1.09938500 -0.00001000 C 1.37960600 0.19046400 0.00000600 C 0.52483300 1.28976400 0.00001200 C -0.85471700 1.09935300 -0.00001900 H -2.45522900 -0.33907200 -0.00000900 H -0.93398400 -2.29559700 -0.00002300 H 1.52111800 -1.95666900 -0.00005300 H 2.45523900 0.33898800 -0.00002300 H 0.93391200 2.29562500 0.00002700 H -1.52105300 1.95672100 -0.00001800 !上面特意是一行空白行 |
因为中性分子得到一个电子后电荷为-1、自旋多重度为2,所以第7行的电荷与自旋多重度分别设为-1与2。
2.1.3 第3步:数据分析
依照上述给出的公式:
VIP = ΔE(cation) – ΔE(neutral) = 214.5kcal/mol
VEA =ΔE(neutral) – ΔE(anion) = -33.9kcal/mol
其中VIP是垂直电离势,VEA是垂直亲和势,ΔE(cation)是阳离子的电子能,ΔE(anion)是阴离子的电子能,ΔE(neutral) 是中性分子的电子能。
2.2 计算苯分子的绝热电离势和绝热亲和势
2.2.1 第一步:优化中性苯分子结构
同2.1.1。
2.2.2 第二步:分别优化中性苯分子失去一个电子和得到一个电子后形成的离子结构
优化中性苯分子失去一个电子后形成的阳离子结构的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 | %nprocshared=8 %mem=1GB # apfd/6-311+G(d,p) opt freq Ph-cation 1 2 C 0.68464900 -1.16997900 -0.00000700 C 1.43481100 0.08738200 -0.00000400 C 0.76538500 1.20836800 0.00000900 C -0.68466200 1.16997800 -0.00000600 C -1.43481200 -0.08738000 -0.00000400 C -0.76537100 -1.20836900 0.00001100 H 1.23649300 -2.10896700 -0.00000800 H 2.51886200 0.04344600 -0.00001400 H 1.25073500 2.17717200 0.00003600 H -1.23648000 2.10898100 -0.00001900 H -2.51886300 -0.04347600 -0.00000800 H -1.25074900 -2.17715900 0.00002100 !上面特意是一行空白行 !第7行为1 2是因为中性分子失去一个电子后的电荷与自旋多重度分别为1与2。 |
优化中性苯分子得到一个电子后形成的阴离子结构的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 | %nprocshared=8 %mem=1GB # apfd/6-311+G(d,p) opt freq Ph-anion -1 2 C -0.44991508 0.72156196 0.00000000 C 0.94524492 0.72156196 0.00000000 C 1.64278292 1.92931296 0.00000000 C 0.94512892 3.13782196 -0.00119900 C -0.44969608 3.13774396 -0.00167800 C -1.14729708 1.92953796 -0.00068200 H -0.99967408 -0.23075504 0.00045000 H 1.49475292 -0.23095104 0.00131500 H 2.74246292 1.92939296 0.00063400 H 1.49532892 4.08996496 -0.00125800 H -0.99981808 4.09002496 -0.00263100 H -2.24690108 1.92972096 -0.00086200 !上面特意是一行空白行 !第7行为-1 2是因为中性分子得到一个电子后的电荷与自旋多重度分别为-1与2。 |
2.2.3 第3步:数据分析
依照上述给出的公式:
AIP = ΔEtherm(cation) – ΔEtherm(neutral) = 209.7kcal/mol
AEA =ΔEtherm(neutral) – ΔEtherm(anion) = -25.8kcal/mol
其中AIP是绝热电离势,AEA是绝热亲和势,ΔEtherm (cation)是阳离子的热校正电子能,ΔEtherm(anion)是阴离子的热校正电子能,ΔEtherm (neutral) 是中性分子的热校正电子能。
热校正电子能可以在输出文件中获得, 以芳基阳离子的热校正电子能为例,下面第6行(Sum of electronic and thermal Energies行)既是:
1 2 3 4 5 6 7 8 9 | Zero-point correction= 0.097910 (Hartree/Particle) Thermal correction to Energy= 0.103055 Thermal correction to Enthalpy= 0.103999 Thermal correction to Gibbs Free Energy= 0.069223 Sum of electronic and zero-point Energies= -231.669807 Sum of electronic and thermal Energies= -231.664661 Sum of electronic and thermal Enthalpies= -231.663717 Sum of electronic and thermal Free Energies= -231.698494 !第6行为热校正电子能 |
关于方法与基组
本文用的apfd/6-311+G(d,p)是示例目的,文献中多用m062x或wb97xd,请读者自行根据需要使用。


