
摘要:目前大多数计算研究还局限在理解现有的实验现象上,将计算化学作为一个手段,在研究初期提供定量的指导还并不多见,为了探究这种可能性,我们介绍了一篇2019年Blanchard课题组发表在JACS上的工作,我们将采用一种问题导向的策略逐步趋向目标的实现,并阐明如何利用理论计算实现一个反应的设计:嘧啶与炔烃的环加成反应。
作者:陈宇 2020-01-13
原文:Le Fouler V, Chen Y, Gandon V, et al. Activating Pyrimidines by Pre-distortion for the General Synthesis of 7-Aza-indazoles from 2-Hydrazonylpyrimidines via Intramolecular Diels–Alder Reactions. J Am Chem Soc. 2019;141(40):15901-15909.

前言
传统的合成化学研究往往是经验主导的,研究者通过文献调研,经验的积累尝试发现新的合成策略,或者拓展已有的策略,这个过程涉及的“反应可能性”的推断往往是定性的,甚至伴随着一些直觉,这种方法的缺点是显而易见的,那么是否有可能通过计算化学理性地设计反应呢?或者退一步说,在现阶段,我们在多大程度上能够通过理论计算在研究的初始阶段对反应的设计提供有价值的指导。值得欣喜的是,量子化学的快速发展已经使理论计算结果能够较为准确地反映出化学过程的关键性质,比如在材料领域,模拟材料的性质;在有机合成领域,理解反应的机理,确定反应的关键步骤等等。然而目前,大多数的计算研究还都是局限在理解现有的实验现象上。将计算化学作为一个手段,在研究初期提供一个定量的指导还并不多见,在这里,我们将尝试探索这一可能性,通过一个例子着重探讨如何利用计算化学设计反应。
在这个案例中,我们将介绍一篇2019年Blanchard课题组发表在JACS上的工作。我们在这里采用一种问题导向的策略逐步趋向目标的实现,并阐明如何利用理论计算实现一个反应的设计:嘧啶与炔烃的环加成反应。
Diels-Alder反应是构建六元环结构重要的策略之一,丰富更多类型的环加成反应将对构建许多有用的环状分子提供重要的基础,尤其在药物分子、天然产物合成以及化学生物学等领域,许多类型的环加成已经被报道,然而嘧啶与炔烃的环加成在此之前却鲜见报道,我们首先构建这个初始模型环加成反应的势能面,如下:

图1. 初始模型环加成反应的势能面
可以看到,反应能垒高达50.0 kcal/mol,说明这个反应在温和条件下很难发生,这也说明了为什么之前报道的这种类型反应必须要在严苛的条件下才能进行,接下来我们将考虑如何使这类反应在较温和的条件下发生。从计算化学的角度讲,我们的策略是:通过对反应分子的改造降低反应能垒(通常我们认为反应能垒在21 kcal/mol之下,反应就能够在常温条件下发生)。
1. 降低“熵效应”的影响
在这个案例中,我们的目的在于实现嘧啶与炔烃模块的温和反应,首先注意到上述的模型反应是分子间反应,也就是说当处于过渡态时,两个独立的分子会被束缚在一起,这个过程是熵减少的过程,由此而带来的自由能升高,我们称之为“熵效应”,为了减少熵效应的影响,我们考虑将分子间反应改造为单分子内反应,也就是将两个反应模块连接在一起。考虑到在过渡态结构中五元环和六元环较为稳定,我们优先寻找能够连接为五元环和六元环的连接部分(如下图2)。
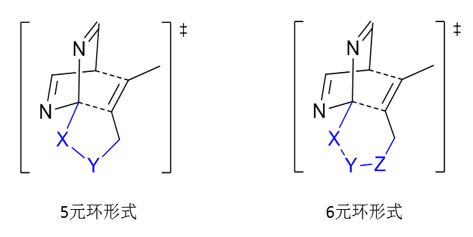
图2. 过渡态结构中五元环和六元环
在这里我们只给出我们筛选成功的例子,感兴趣的读者可以尝试其他的连接方式。图3是我们第一次分子改造之后的势能面。

图3. 改造后的势能面
可以看出尽管反应总能垒仍然高达48.8 kcal/mol,但反应的单步能垒(从Pre-B到TS-B)已经降到了39 kcal/mol。注意到总反应能垒中,有9.4 kcal/mol来自于反应物B到Pre-B的结构变化,之所以要先发生一步构象变化,是因为能量最低的构象并不直接与过渡态相连,为了达到过渡态的结构,分子需要先经过一步构象变化以达到过渡态结构,这个过程也称为分子的预重构。那么接下来我们采用什么策略呢?
2.降低结构重构化能量
从图3可以看出,预重构的过程需要9.4 kcal/mol,因此我们考虑第二步降低预重构过程能量,对分子B和Pre-B的结构进行分析可以看出,分子B的共轭特性是其比Pre-B更稳定的根本原因,因此我们的改造方向集中在破坏分子B的共轭特性,一个方法便是将H原子用其他更大的取代基替换(如下图4)。
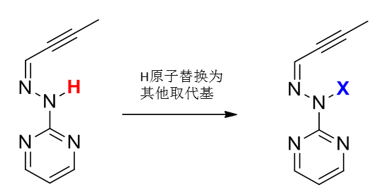
图4.H原子用其他更大的取代基替换
我们直接给出筛选出的有较好结果的取代基,读者也可以尝试其他不同的类型。势能面如下图5:
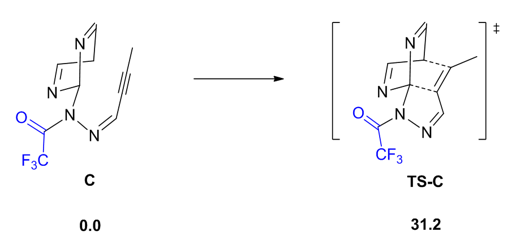
图5. 有较好结果的取代基的势能面
可以看出图5中当破坏了分子的大共轭后,原来的预重构结构反而成了能量最低的构象,同时由于取代基的电子效应,反应能垒降到了31.2 kcal/mol,相比于模型反应的50 kcal/mol降低了接近20 kcal/mol。
3.评估取代基电子效应
一个策略的优劣有一些基本的标准,反应底物的广度便是其一,能够适用于更多的反应物显然更有优势,因此我们也可以评估不同取代基的电子效应对反应活性的影响,下图6是我们筛选的可以替换不同取代基的位点(X和Y位点)。
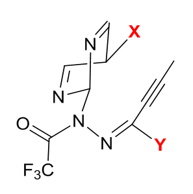
图6. 可以替换不同取代基的位点(X和Y位点)
读者可以自己尝试一下不同取代基的效应。
在这个案例中,我们经过三次改造,便在能量的评估上达到了较好的效果,实验结果也与计算结果相一致。我们演示了如何利用计算化学通过“分析→改造→反馈”的流程循环对反应进行改进,以达到我们最终的目的。
总结
这里演示的方法的仅仅是众多设计方法中的一种,为了使某类反应能够发生,我们还可以设计催化剂,重要的是先要明确研究的目标,再利用基本的物理化学规则,通过不断地改造→反馈→再改造,最终实现相应的目的。
参考文献
- Le Fouler V, Chen Y, Gandon V, et al. Activating Pyrimidines by Pre-distortion for the General Synthesis of 7-Aza-indazoles from 2-Hydrazonylpyrimidines via Intramolecular Diels–Alder Reactions. J Am Chem Soc. 2019;141(40):15901-15909. doi:10.1021/jacs.9b07037


