
文献重现 | 大环EGFR激酶抑制剂BI-4020的先导化合物5与6的发现与设计
摘要:本文重现了Boehringer Ingelheim的科学家在设计大环EGFR激酶抑制剂BI-4020时先导化合物5与6发现的过程。演示了如何用Flare QM进行扭转角分析(Torsion Profile)以确定优势构象;用SPARK以开环化合物5为起点设计大环化合物6;并用FreeForm评估了化合物5与6的构象稳定性。
肖高铿/2020-01-18
背景
当携带诸如del19或L858R之类的激活突变时,表皮生长因子受体(EGFR)在一部分肺肿瘤中起致癌作用。尽管肿瘤对酪氨酸激酶抑制剂(TKIs)的反应伴随着明显的肿瘤缩小,但这种反应通常并不持久。通常由于EGFR激酶结构域突变而获得了TKI耐药性,大多数患者在治疗后两年内复发。至关重要的是,目前批准的EGFR TKI不能再抑制同时具有T790M和C797S两种抗性突变的致癌EGFR。
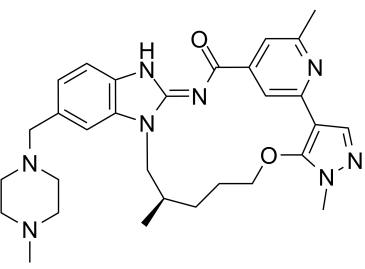
Figure 1. 大环EGFR激酶抑制剂BI-4020
BI-4020是一种第四代、具有口服活性、非共价的EGFR酪氨酸激酶抑制剂。BI-4020不仅抑制三联突变的EGFRdel19 T790M C797S突变体(在 BaF3 细胞系中,IC50=0.2 nM),也抑制双重突变的EGFRdel19 T790M和单突变的EGFRdel19(IC50=1 nM)。BI-4020 同时还保留了对野生型EGFRwt的活性 (IC50=190 nM)。BI-4020 显示高激酶组选择性和良好的 DMPK特性[1]。
Engelhardt等人[1]报道了大环激酶抑制剂BI-4020的发现过程,其关键点是:通过筛选激酶抑制剂数据库发现高选择性但中等活性的苯并咪唑类化合物,然后通过大环化将分子完全刚性化。
QM-扭转角特征分析(QM-Torsion Profile)与BI-4020先导化合物5的发现
先导化合物5的发现

Figure 2. 化合物1与ATP结合位点的相互作用(PDB 6S9B)
苯并咪唑化合物1是筛选到的苗头化合物,如图2所示,其与EGFRL858R T790M的共晶结构(PDB:6S9B)形成了双点铰链结合:苯并咪唑环上N-H、羰基与EGFR铰链区MET793发生氢键结合。异丁基羟基填充了激酶的糖结合口袋,而R2-位取代吡啶(的N)与口袋里磷酸结合区的LYS745形成氢键。Met790 gatekeeper残基与苯并咪唑没有发现立体碰撞。进一步的优化可以发现化合物1两个相连芳环的扭转角(torsion angle)约为40°,量子力学-扭转角扫描(QM-torsion angle scan)计算表明该角度是一低能构象(见图3)。
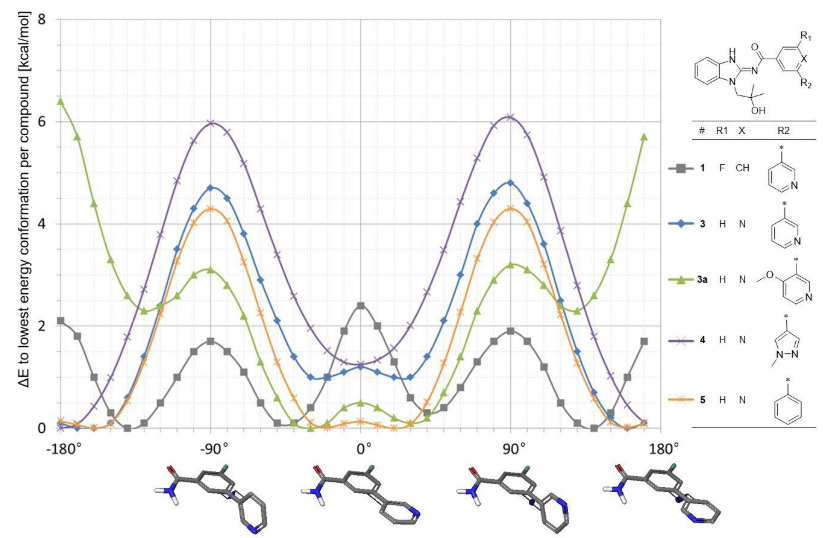
Figure 3. 化合物1,3,3a,4与5的量子化学-扭转角扫描
苯环R1-位当去掉F原子后的化合物2,在生化与生物标记物活性测试中与化合物1具有相似的活性,但更易于合成,故后续优化不在苯环R1-位进行取代。
在着手解决Thr854侧链羟基与芳香环系中心X-位N受体的相互作用。新设计的化合物应当保留低能构象且以30-40°的夹角与Lys754相互作用。为了发现此类化合物,对化合物3、3a与4的双芳环系进行量子力学扭转角扫描(见图3)。
与化合物1相比,化合物3的R2片段上的吡啶N使得联芳系进一步去对称化,造成30°与160°夹角的两个极小点之间的能量差变地更大(图3)。比之两个N在异侧的160°极小点(其偶极矩有部分相互抵消),在30°极小点时化合物3的两个吡啶N在同侧导致不利的偶极-偶极相互作用(偶极矩:3.87 Debye)。
化合物3的30°极小点使配体能够通过R2片段上的吡啶N与Lys745发生相互作用。然而,这个极小点比全局极小点(160°夹角)能量高约1kcal/mol。因此,需要对这个非优选的两面角支付出一定的能量惩罚。但是,结合亲和力的获益是由于与Thr854的相互作用。令人感兴趣的是,化合物3在酶学水平呈现出3-8倍的活性提高,并在细胞水平上呈现出显著的活性增强,同时对野生型EGFR保留有低亲和力的活性。
对于化合物4,观察到类似情况。 在0°的扭转角处发现了一个局部最小值,而绝对最小点(位于180°)则降低了约1kcal/mol。同样,必须为非最佳二面角支付能量损失。但另一方面,由于与Thr854的相互作用可以预期获得结合亲和力。 同样,对于化合物4,在酶学水平观察到活性略有改善,并且在细胞水平甚至观察到了更明显的活性增强。
针对R2位研究了几种不同的芳香环体系,目的是识别出与EGFR相互作用时的联芳基二面角为绝对能量最小点。化合物3a是一种可能的解决方案。该化合物在R2位有4-甲氧基-吡啶-3-基部分。联芳基体系的QM扭转角扫描(见图3)显示,由于甲氧基与对面的吡啶基N的不利相互作用,二面角为能量很高的160-180°,但在30°时为绝对最小点。对于化合物3a,两个环氮原子在30°时具有不利的偶极-偶极相互作用,但能量损失被甲氧基与面对面的质子有利静电相互作用所补偿(参见补充图1)。因此,配体3a在30°的能量比化合物3低。与配体3和4相比,配体3a的这种优化的结合构象使其对EGFRL858R T790M C797S的酶学活性提高了30倍。EGFRdel19 T790M C797S的酶学活性数据也有同样的趋势。与配体3和4相比,这种改善的酶学活性可导致p-EGFRdel19 T790M C797S细胞水平活性提高35倍,抗增殖作用提高5倍。但是,与化合物3和4相比,该化合物的激酶选择性明显较低(命中率为36%;请参见表1)。这些数据表明,通过与Lys745的最佳相互作用可以大大的提高活性,但以降低激酶选择性为代价。
作者假设,如果Lys745(磷酸区域)周围的口袋被不带受体的芳香族体系所占据,则可以获得更好的激酶选择性。 该策略的先决条件是预想的R2部分不会与Lys745发生立体冲突,这可以通过小于20°的二面角来实现。
表1. 化合物1-5的SAR
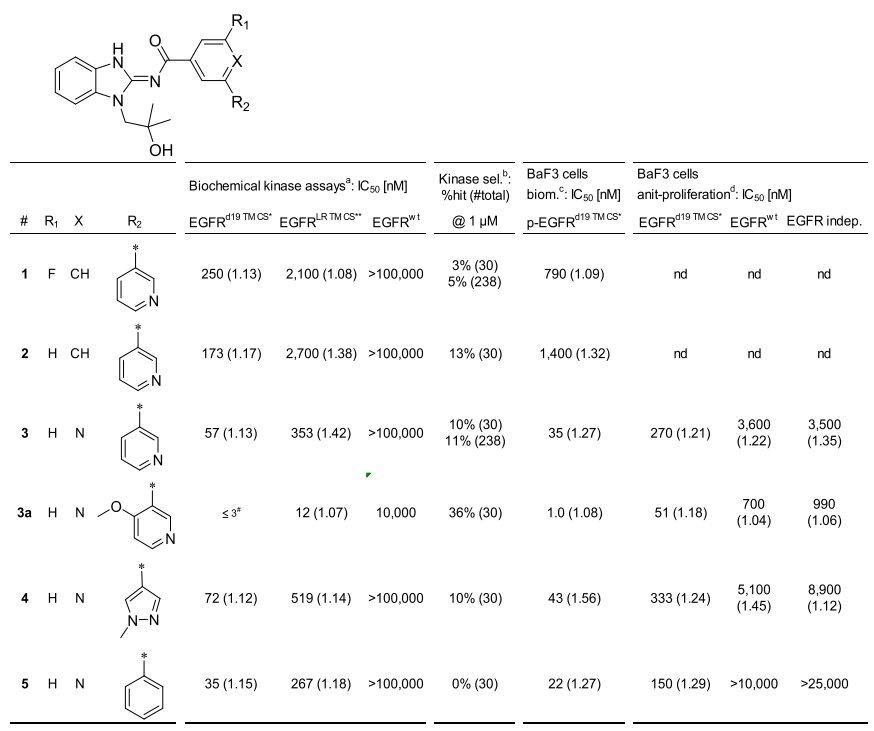
为了验证该假设,他们合成了苯并咪唑化合物5,该苯并咪唑化合物5在第二个芳环中不包含氢键受体原子,并且在R2位仅携带对称的苯环。因此,在最小点之间(夹角20°和160°,见图3)几乎看不到能量差。令人高兴的是,尽管与化合物3相比,对EGFR突变细胞的酶学与细胞水平的活性基本没有变化,选择性却得到了显着提高,对非EGFR依赖的细胞毒活性实验可以证明这一点。同时,发现激酶选择性非常高,在一组30种激酶的测试中仅命中突变的EGFR。 此外,比较BaF3细胞系EGFRdel19 T790M C797S与EGFRwt的抗增殖作用可以发现,苯并咪唑类化合物5对野生型-EGFR缺乏抑制作用(参见表1)。
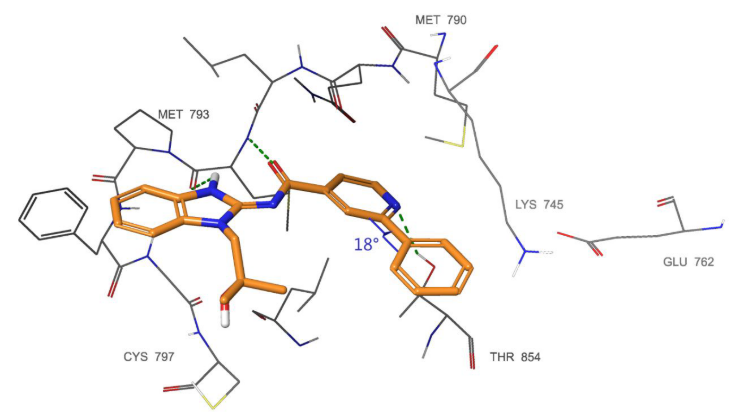
Figure 4. 化合物5与EGFRL858R T790M的相互作用(PDB 6S9C)
化合物5与EGFRL858R T790M的复合物晶体结构表明(见图4),该化合物以预期的大约20°的联芳基二面角与蛋白质结合。此外,与Lys745和Glu762的距离足够大(3.5Å)可以避免发生碰撞。
QM扭转角分析(Torsion Profile)
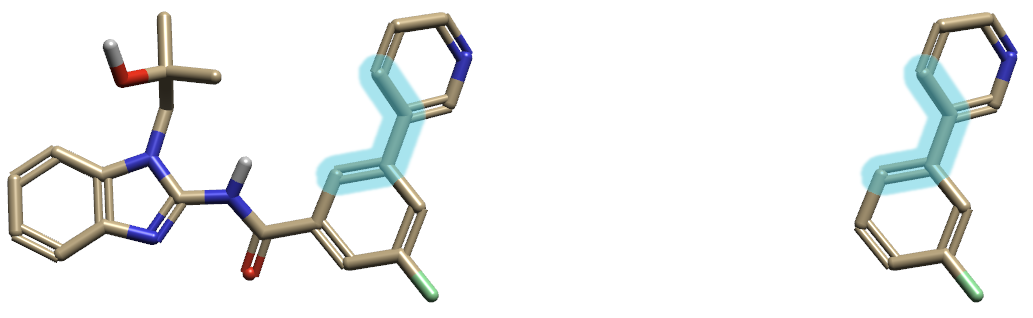
Figure 5. 左:化合物1(PDB 6S9B的配体),右:化合物1的模型分子。高亮原子:QM扭转角分析的两面角
在先导化合物5的发现过程中,大量地应用量子力学扭转角分析以发现优势的构象。以化合物1的联芳基部分(图5)为例,为了加快计算并取得高可靠性的结果,用模型化合物(图5-右)代替化合物1进行扭转角分析。扭转角分析采用Flare QM的Torsion Profile2在R2SCAN-3c/DEF2-mTZVPP理论水平下进行计算,定义的两面角见图5的高亮原子,主要参数如下:
- Method: DFT
- DFT functional: R2SCAN-3C
- Use dispersion correction:Yes
- Basis set: def2-mtzvpp
- Convergence: Medium
- Max iterations: 500
- Number of rotomers: 72
- Degrees: 5.0°
- Max Threads: Auto
其中Degrees是根据Number of rotomers自动生成,无需主动设置,这里意味着以5.0°为步长进行两面角扫描。
结果如图6所示,可以发现生物活性构象就是最低能构象40°,这与原文报道的一致,化合物1联芳基片段的生物活性构象是稳定的。
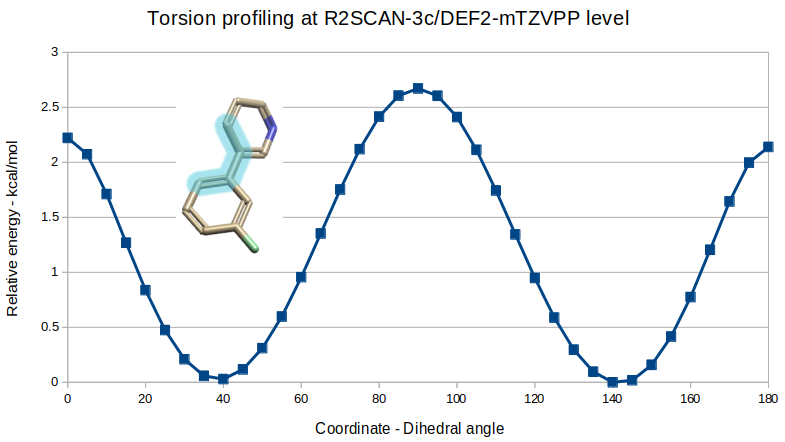
Figure 6. 化合物1的QM扭转角特征分析(QM-Torsion profile)结果
大环EGFR激酶抑制剂的设计
化合物5代表了具有选择性但是活性强度不足的化合物,作者借鉴了lorlatinib的设计思路[4]:通过大环化来降低配体的熵变以提高化合物的生物活性,秉承理论与实验相结合思路进行大环分子设计。二维核磁(2D-NMR)实验与DFT计算的玻尔兹曼权重表明,化合物5具有两种优势构象:活性与非活性构象。因此假设如果可以将化合物5约束在活性构象则可以提高其对EGFR的结合亲和力。
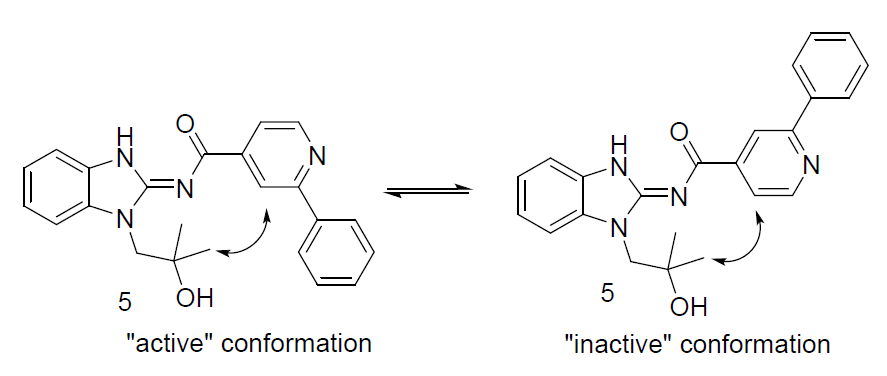
作者以化合物5为起点化合物(starter),设计了构象约束的大环化合物6:比起开环化合物,大环化合物可能活性得以提高。然而,这里面有两个问题要回答:1)有什么样的大环化合物可能满足预期的构象约束效果?2)鉴于大环化合物比开环化合物更难合成,所以需要考虑感兴趣的大环化合物是否值得去合成(真地具有更低的张力能以提高结合亲和力)?
以化合物5为起始化合物设计大环化合物
第一个问题可以用SPARK来回答,之前已经演示过如何用SPARK设计大环BRD4抑制剂[3]。与BRD4抑制剂的大环化类似,详细的操作过程请参见视频1。在该视频中,用SPARK重现了Engelhardt等人[1]以化合物5(PDB 6s9c)为起始分子,设计大环分子6(PDB 6s9d)的过程。
视频1. SPARK演示:以化合物5起点的大环化合物设计
大环化设计需要指定两个进行连接的区域,以PDB 6S9C中化合物5为起始化合物,如下图7所示,设定了需要被链接起来的两个区域。化合物5该区域的原子将被替换,新片段从开始生长。

Figure 7. 以化合物5为起始化合物,设定两个需要被连接进行大环化的设计
还规定了连接点1(2-羟丙基部分)的原子类型必须是sp3的C,而连接点2(苯环上的H)的原子类型必须为sp3的O。并将2-羟丙基部分羟基形成的两个场点(图8箭头指示部分)移除。
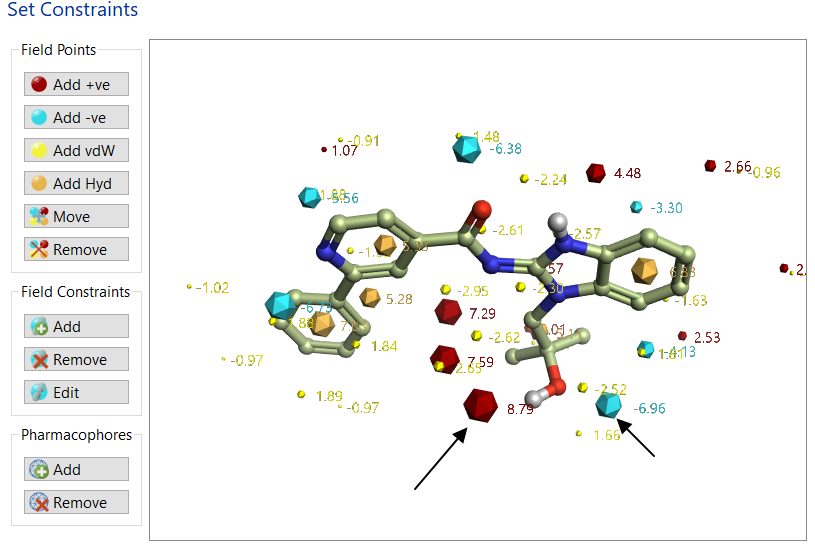
Figure 8. 设置约束条件的时候,移除了羟基的两个场点
在该演示中SPARK给出了21个含-CCCCCO-连接的大环化合物。图9展示了其中部分化合物,你会发现化合物6也被SPARK设计出来了,其中有的化合物还被甲基取代。
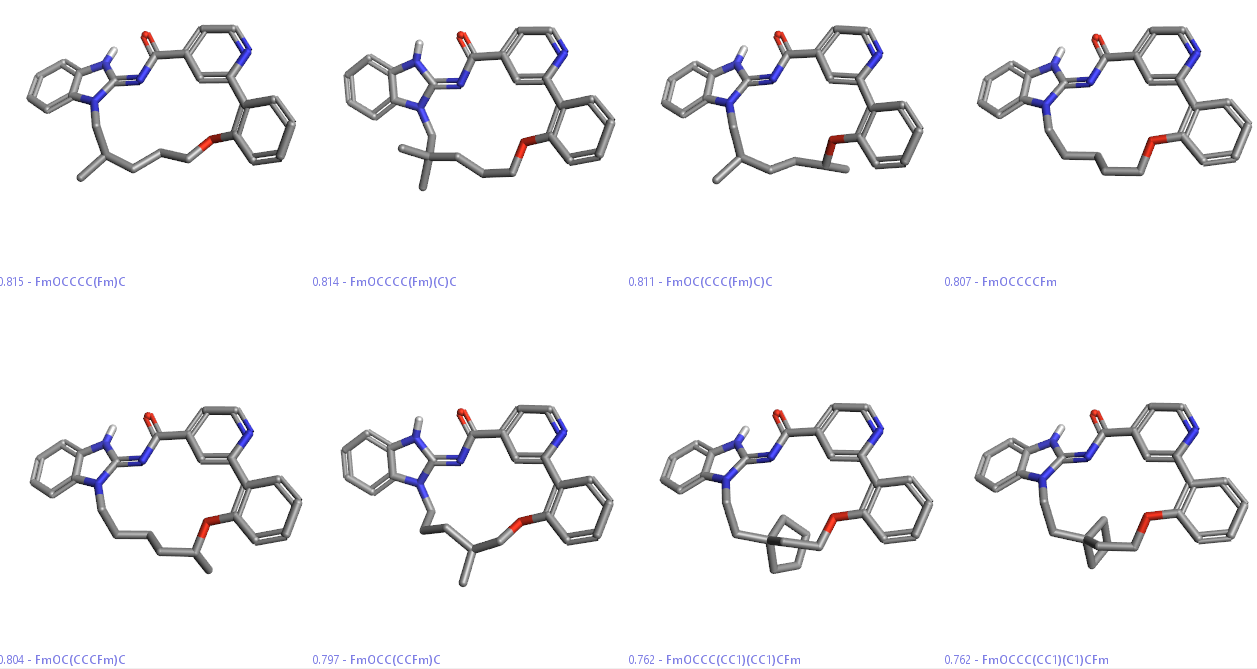
Figure 9. 部分与化合物6同样链长的大环化合物,化合物6(右上角)也被SPARK设计出来
应该注意到,SPARK给出了非常多有意思的大环化合物。比如还有含6个C的-CCCCCCO-连接的大环化合物,以及醚类、酯类、烯烃等等。图10展示了6个-C6O-连接的化合物
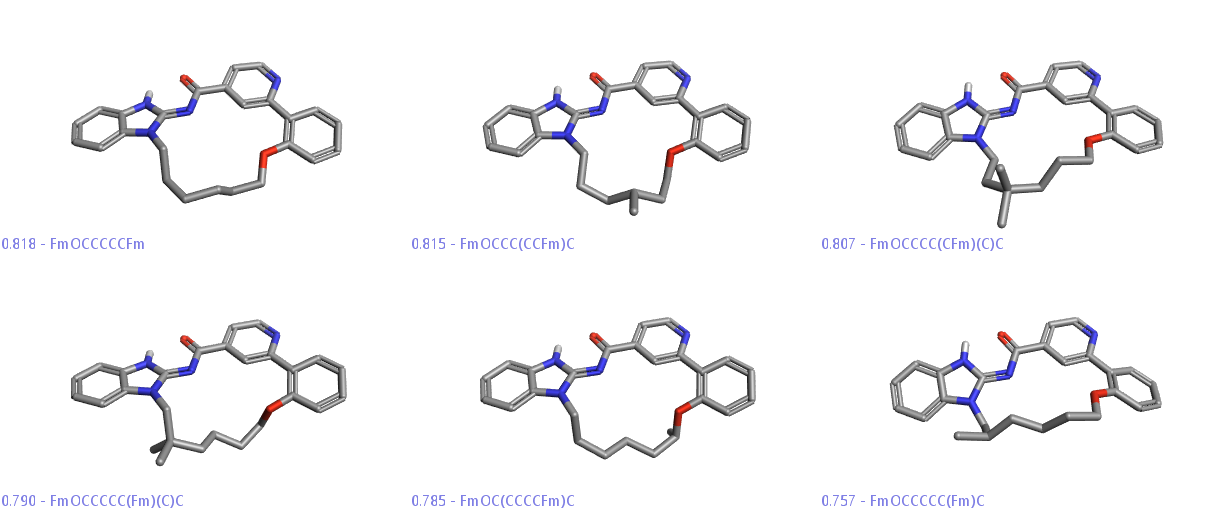
Figure 10. 6个包含CCCCCCO-的大环化合物
图11展示部分连接臂为醚类或酯类的大环化合物。
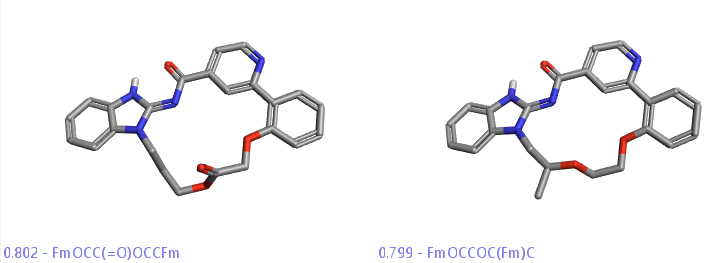
Figure 11. 连接臂为醚类与酯类化合物
构象稳定性评估:该化合物是否值得合成?
如前所述,大环化是为了稳定构象使得熵变对结合有利。但是熵变并不直观,通常需要计算才能准确评估。如何系统地评估一个结合构象的稳定性,我们在FreeForm–构象稳定性评估及其在先导化合物优化中的应用[4]一文里进行了详细的介绍。从化合物5到6的大环化设计并没有增加或减少相互作用,因此很适合用FreeForm来评估活性差异。在这里,我们分别从PDB 6S9C与6S9D提取了化合物5与6的生物活性构象,用FreeForm计算了全局张力能,结果分别见图12与13,并总结在表2中。
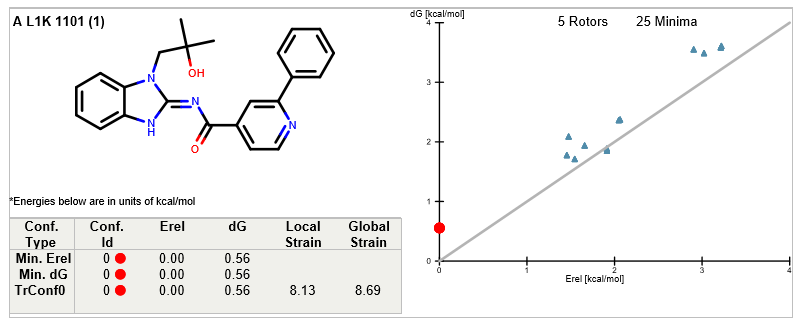
图12. 化合物5的全局张力能计算
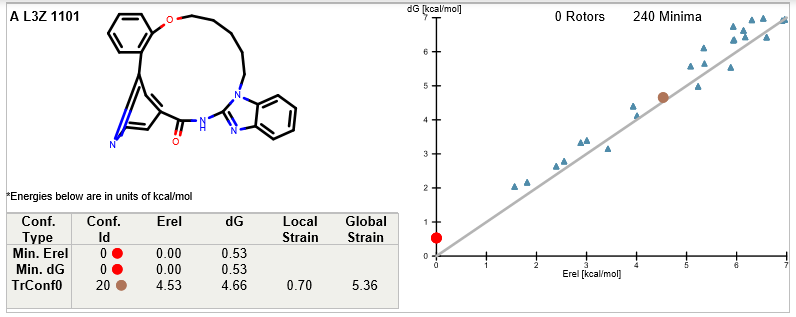
图13. 化合物6的全局张力能计算
化合物5与6的全局张力能计算总结见表2,可以发现:开环化合物5比大环化合物6在局部极小点自由能(Relaxed bioactive conformer ΔG)占优势(分别为0.56与4.66kcal/mol);但是在Local ΔH远远地处于弱势(化合物5,6分别为8.13与0.70kcal/mol)。也就是开环化合物5需要比大环化合物6花更大的能量去维持活性构象。这导致化合物5比化合物6具有更高的全局张力能(Global Strain ΔE),分别为8.69与5.36kcal/mol。总的来说,在大环化过程中没有增加或减少相互作用的情况下,全局张力能与化合物的活性的直接相关。从配体的全局张力能角度讲,化合物6比5更有优势,大环化合物6值得一试,这与酶学IC50的实验结果一致。
表2. 化合物5与6的全局张力能比较
| Items | 5 | 6 |
|---|---|---|
| Relaxed Bioactive ΔG | 0.56 kcal/mol | 4.66 kcal/mol |
| Local ΔH | 8.13 kcal/mol | 0.70 kcal/mol |
| Global Strain ΔE | 8.69 kcal/mol | 5.36 kcal/mol |
| IC50 | 267 nM | 16 nM |
小结
本部分我们用SPARK实现了从开环化合物5出发,设计了一系列大环化合物,其中包括化合物6以及其它有意思的系列化合物,重现了Engelhardt等人先导化合物6的发现过程。并用OpenEye/FreeForm对大环化合物6与开环化合5进行了构象稳定性评估,结果表明大环化合物6比开环化合物5具有更低的全局张力能,与体外的酶学实验结果一致。
文献
- Engelhardt, H.; Böse, D.; Petronczki, M.; Scharn, D.; Bader, G.; Baum, A.; Bergner, A.; Chong, E.; Döbel, S.; Egger, G.; et al. Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62 (22), 10272–10293. https://doi.org/10.1021/acs.jmedchem.9b01169.
- Flare. https://www.cresset-group.com/software/flare
- 案例 | 用SPARK大环化技术设计大环BRD4抑制剂. http://blog.molcalx.com.cn/2018/03/09/using-spark-to-design-macrocycle-brd4-inhibitors.html
- FreeForm–构象稳定性评估及其在先导化合物优化中的应用. http://blog.molcalx.com.cn/2016/07/02/freeform-conformer-stability.html
- Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10 R )-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro- 2H -8,4-(metheno)pyrazolo[4,3- h ][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c. J Med Chem. 2014;57(11):4720-4744. doi:10.1021/jm500261q


