
摘要:VCD与ECD是两种重要的立体化学结构确证方法。当化合物在紫外区没有科顿效应时,需要用VCD来进行结构确证。本教程叙述了如何用Gaussian计算柔性化合物的VCD图谱,包括:1)构象搜索生成多构象文件;2)导入构象搜索结构,批量进行VCD计算;3)玻尔兹曼加权生成VCD图谱。
作者:肖高铿
日期:2018-01-09
一. 振动圆二色性谱简介
圆二色性谱(CD)常用于化合物的立体化学结构确证。电子圆二色性谱(ECD)适用于在紫外区有Cotton效应的化合物,而VCD适用于紫外区没有Cotton效应的化合物结构确证。本文介绍了VCD的计算流程,包括:如何生成化合物的构象、对每个构象进行优化与VCD计算、以及对多个构象进行玻尔兹曼加权平均得到最终图谱。
二. 计算流程
VCD的计算流程与ECD一样,需要先对化合物进行构象搜索、对每个构象进行优化、计算VCD、再对每个构象的VCD图谱按玻尔兹曼进行加权平均得到最终的图谱。因此本教程包含了下列步骤:
- 构象搜索
- VCD计算
- 生成玻尔兹曼加权平均的VCD图谱
构象搜索可以采用CONFLEX来进行,其特点在于(1)采用穷尽地构象搜索策略;(2)考虑熵变;(3)可以考虑溶剂效应。因此,采用CONFLEX可以方便对构象进行初步排序,具体操作步参见:《CONFLEX教程 | 构象搜索》
构象搜索还可以用GMMX,GMMX提供了多种策略进行构象搜索,目前与GaussView完全整合作为一个独立模块使用,教程见:GaussView 6 | Help | Tutorial | GMMX Conformer Tutorial。
除了CONFLEX与GMMX之外,用Forge与Torch还可以进行构象搜索,见《Torch教程|构象搜索》,适用于Torch的教程都适用于Forge。
本教程还适用于红外,拉曼,NMR的图谱预测。
三. 具体操作流程
以图1化合物desflurane为例,说明VCD计算的流程。
图1. 算例化合物desflurane
3.1 构象搜索
这里我们准备了一个已经用CONFLEX完成构象搜索的文件:desflurane_conflex.sdf
根据CONFLEX的MMFF94力场、吉布斯自由能的计算,构象分布如下表1所示。
表1. CONFLEX计算结果

从GPOP(根据吉布斯自由能计算的玻尔兹曼分布)列可以看出,前7个构象占据了99.6%的构象空间,用这7个构象计算的性质可以模拟化合物的真实性质。
3.2 VCD计算
-
用GaussView 6读入CONFLEX的构象搜索结果文件
-
选取合适的构象
-
批量结构优化与VCD计算
GaussView File|Open 选取desflurane_conflex.sdf, 点击Open File按钮读入文件。
如图2所示,拉动滚动条,找到TOTAL GIBBS FREE列(步骤1)。点击这个条目,可以对行进行排序,我们让它从低到高排序。按住SHIFT键,用鼠标选中能量最低的7个构象。
注意:在Reading MDL SDF file对话框里有BOLZMANN POPULATION%列,这个构象分布是根据构象能(势能)计算,而不是吉布斯自由能!我们推荐用吉布斯自由能来计算构象分布。
确保Target处为Single new molecule group for all molecules,点击OK按钮,这时被我们选中的7个构象被打开(见图3)。
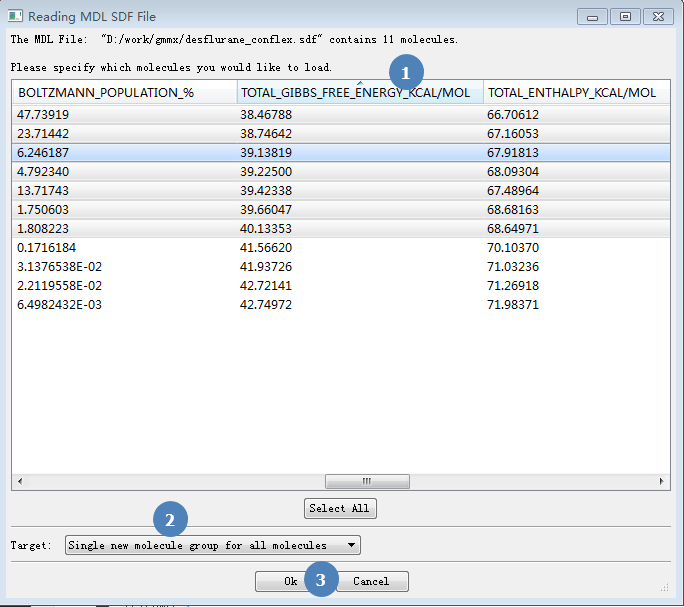
图2. 构象选取
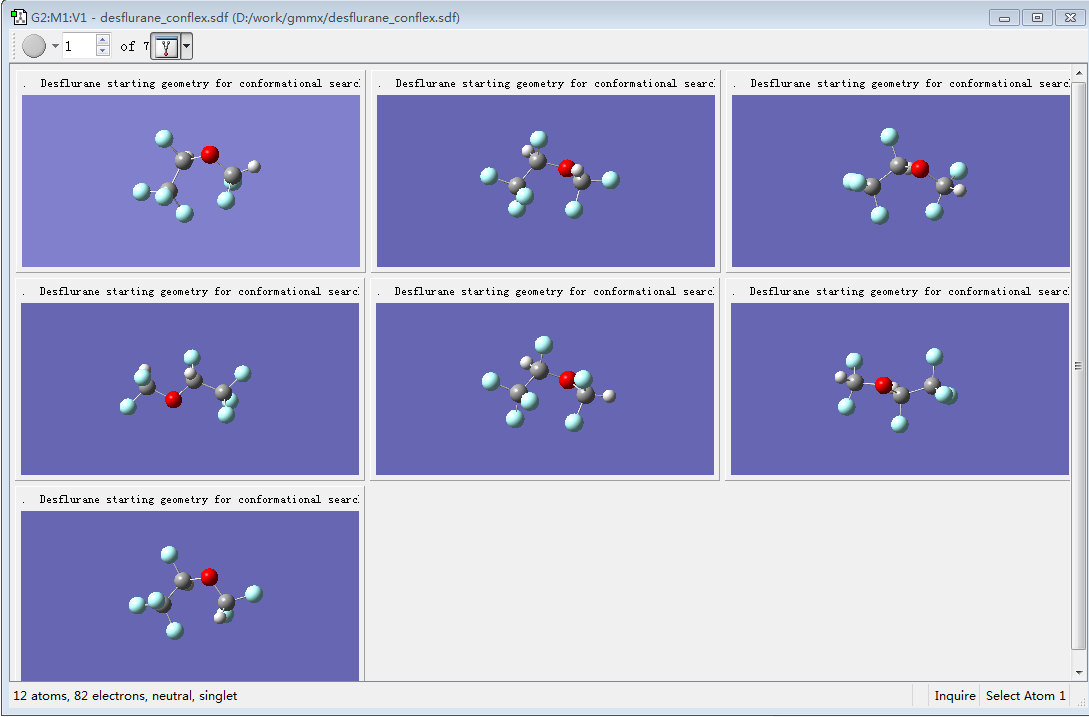
图3. GaussView 6观察desflurane的7个构象
我们现在开始对Desflurane的不同构象进行精确的构象优化与VCD光谱计算。
GaussView | Calculate | Gaussian Calculation Setup
图4. 开始设定Gaussian计算作业
在Job type选项卡里选择作业类型为Opt+freq,表示我们要进行Optimizaiton与Frequency计算,并将Compute VCD按钮打钩表示我们要进行VCD计算。
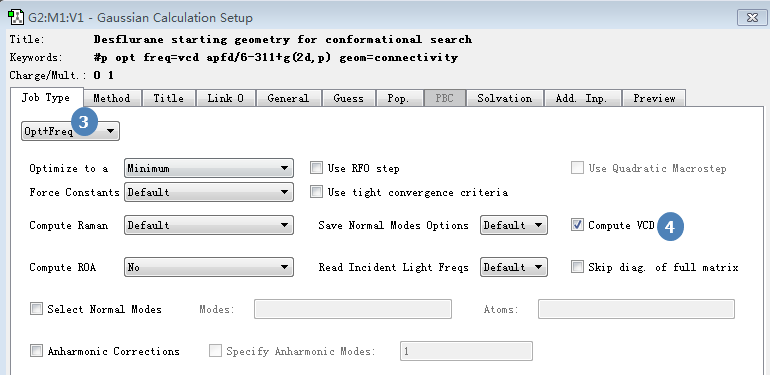
图5. 作业类型的设定
如图6所示,在Method选项卡里确保Method处为Ground state,并且采用DFT在APFD/6-311g(2d,p)理论水平进行计算。
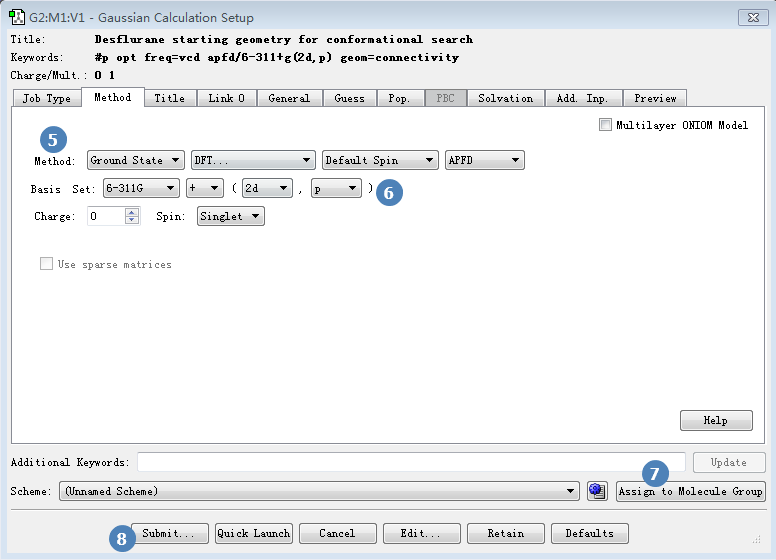
图6. 计算方法的设定APFD/6-311g+(2d,p)
因为我们要对7个构象采用同样的参数进行计算,所以要点击一下右下角的Assign to Molecule Group按钮(图6步骤7);再点击submit按钮,提交作业,会有消息框提示您是否保存作业文件,点击Save按钮,弹出Save Input file对话框。
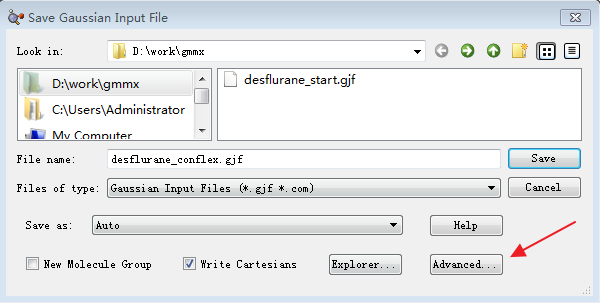
图7. 保存输入文件的Advance按钮
点击Advanced按钮, 会打开Save Structure File对话框,可以让我们一键保存7个构象的Gaussian输入文件。
如图8所示(步骤1),将Save Molecule Group设置为“Yes, separate file for each molecule”。
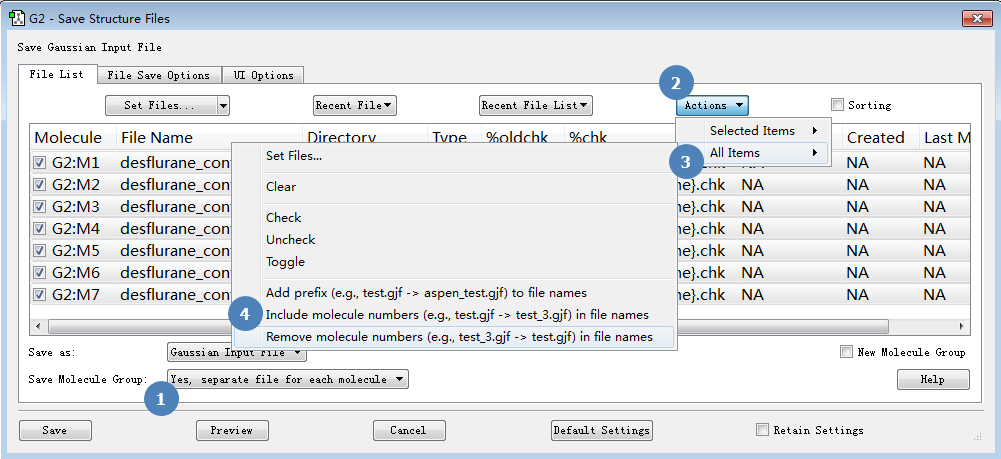
图8. 设置保存文件方法
然后点击选择:Actions | All Items | Include molecule numbers in file names,给每个构象生成一个带”_数字”后缀的输入文件,如图8所示。
如图9所示,确认需要保存的输入文件都被打上“对勾”,点击Save。
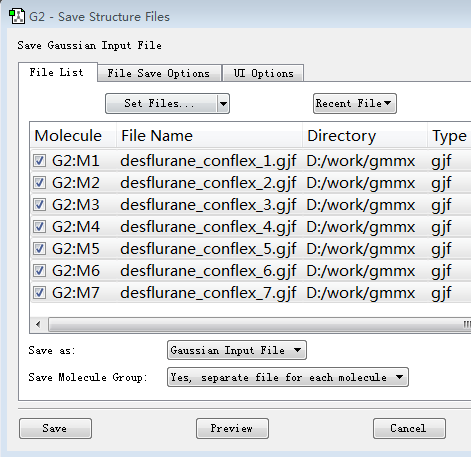
图9. 设置要保存的文件
在Submit job对话框中(图10)勾选“Confirm submission of Jons at SCJonMan”,点击Yes按钮开始计算。
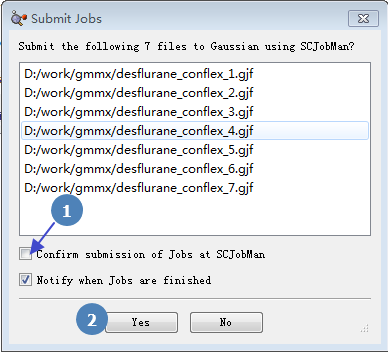
图10.将作业提交给SCJobMan开始计算
3.3 VCD的图谱生成
1. 打开log文件
GaussView | File > Open File 选中生成的7个log文件,注意确保Target下拉选项为“Single New Molecule Group for all files”,如图11所示,点击Open按钮。
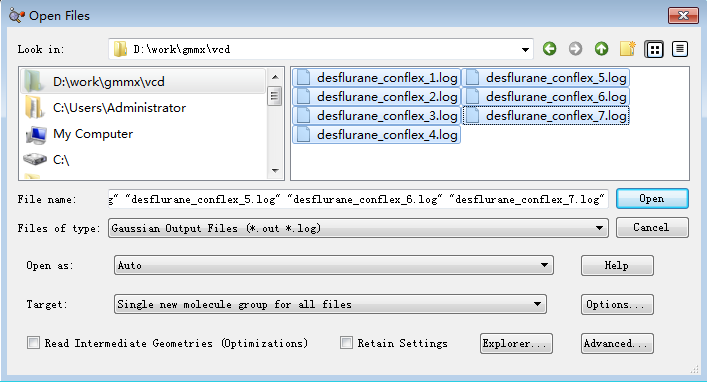
图11. 打开log文件
结果如图12所示,将7个化合物都展示出在视窗里。
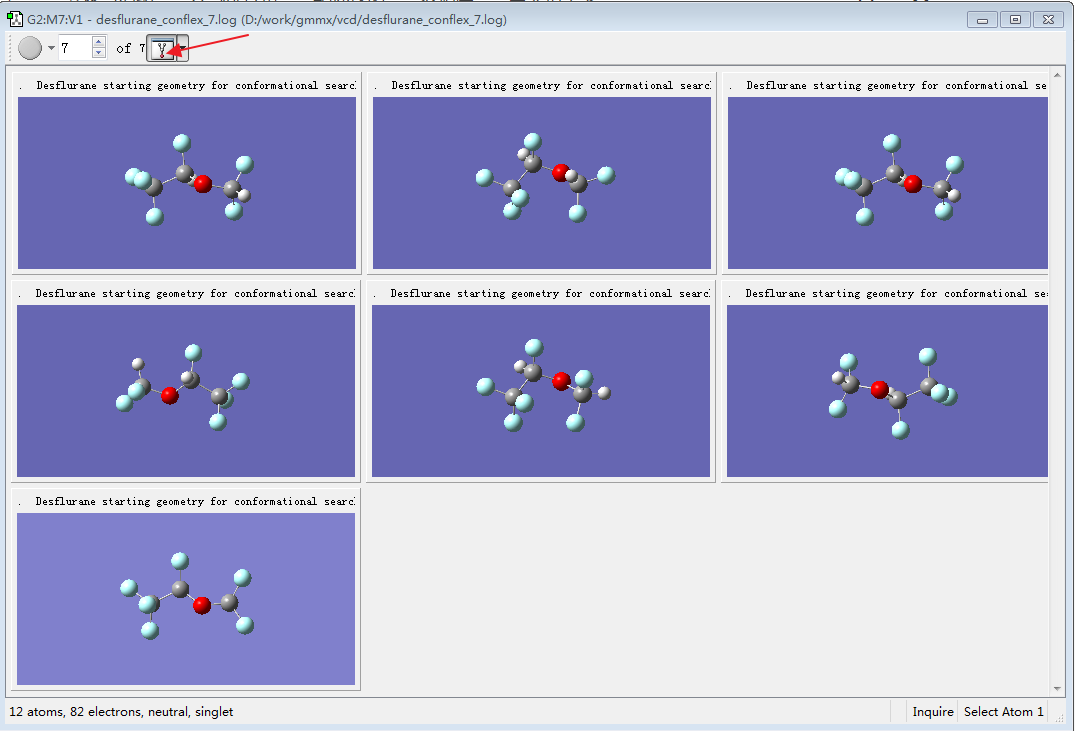
图12. 7个打开的log文件
2.生成图谱
GaussView | Results > Vibrations; 在打开的Vibration窗口里点击Spectra按钮,然后在Vibratiopn Spectra对话框Polt下拉选项里选择Mixture Spectra,如图13所示。为了操作方便,点击Plot下拉选项里的IR,可以隐藏红外图谱而仅展示VCD图谱。
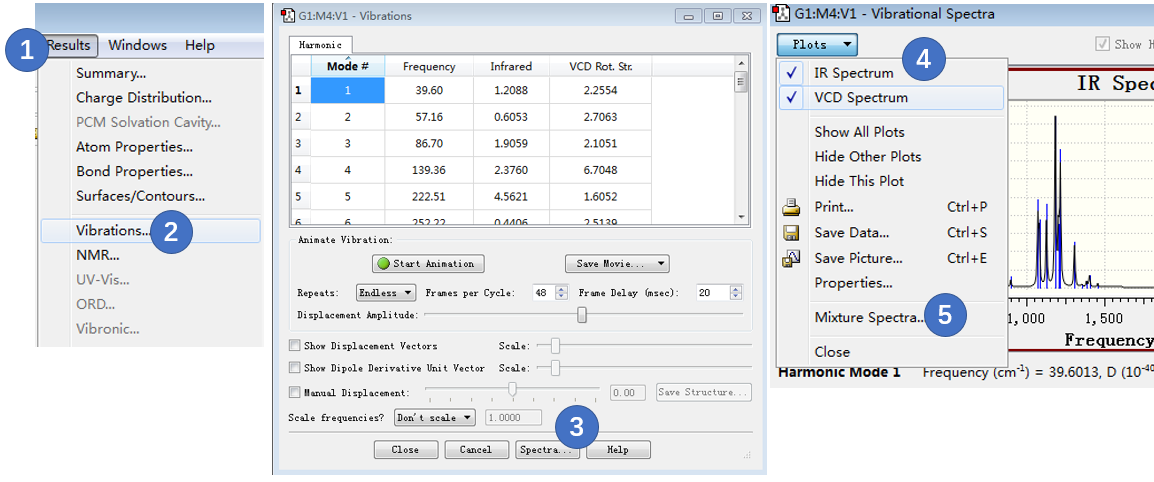
图13. 生成图谱
现在我们可以看到VCD图谱,但这个图谱是多个个构象的图谱叠加在一起的结果,杂乱无章不可解释(图14):既有每个构象的图谱,也有每个峰的棒图。接下来,我们要将单个构象的峰对应的棒图隐藏、仅保留各个构象的VCD图谱以及按Boltzmann加权平均得到最终图谱。
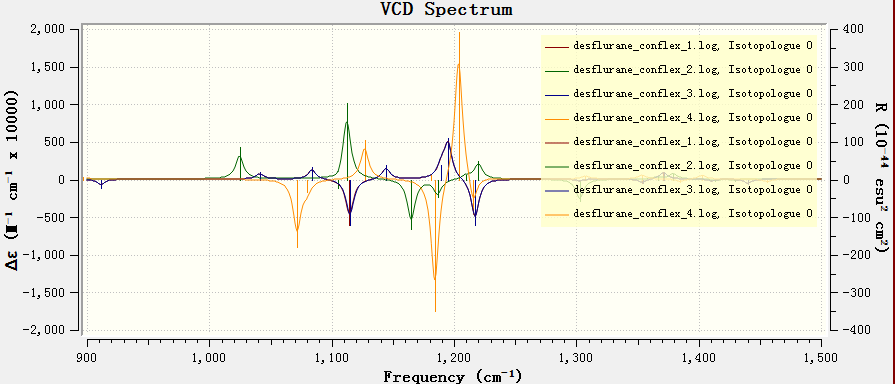
图14. VCD图谱
在Vibration Spectra的Plot下拉菜单里选择Mix Editor设定图谱的展示方式(图15步骤1)。先设定组合图谱(7个构象的玻尔兹曼加权平均图谱)特性:在Combination区,设定Plot Style为Curve(图15步骤2);点击Stick Pen设定Style为None;点击Curve Pen,设定Style为Dash,Width设为2.0px。
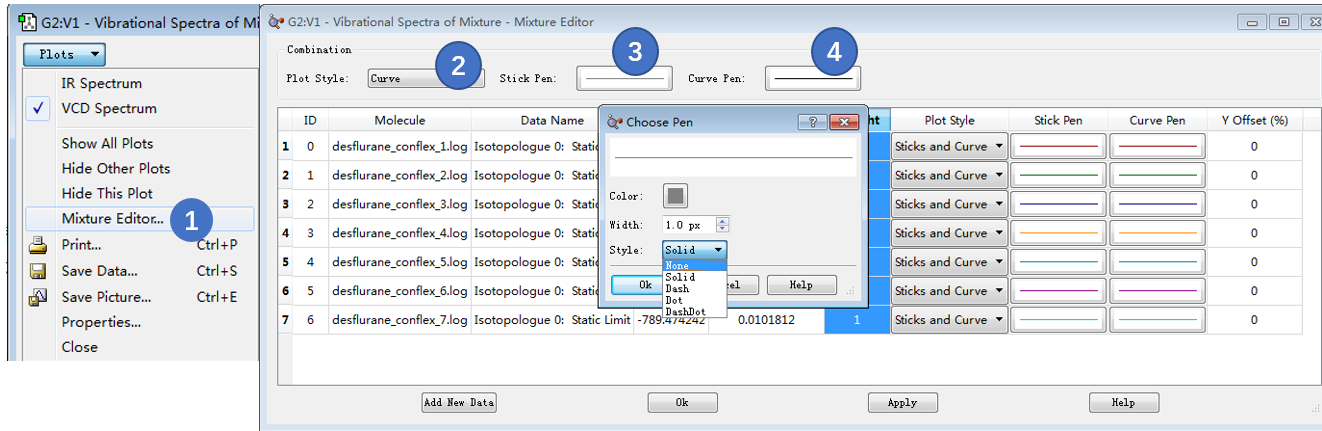
图15. 编辑VCD组合图谱曲线,设定玻尔兹曼图谱的外观为2px宽的虚线
点击标题行Weight处,该列则被选中。将鼠标移植Weight下面的任意一行,点击鼠标右键,选择“Assign Bolzmann Population Weights”。你会发现,每个构象的权重由1变为Bolzmann权重,如图16所示。

图16. 设定每个构象的权重
现在开始设定每个构象的VCD曲线风格,如图17所示,在Plot Style列,单击任意一个格子,选择风格为Curve。依次将每个构象的Plot Style都设为Curve。
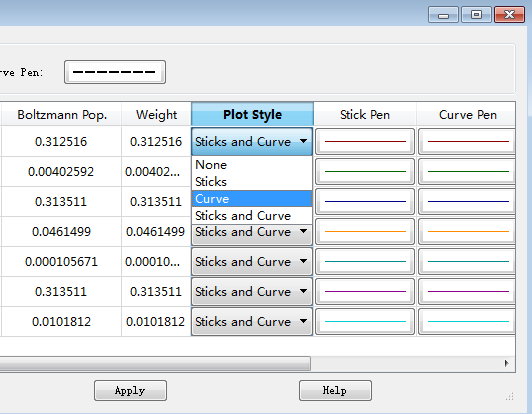
图17. 设定每个构象VCD图谱的风格为Curve
图谱绘制风格设定完毕,点击OK按钮退回VCD图谱界面,在图谱上点击右键,去掉Legend(图例),结果如下图18。
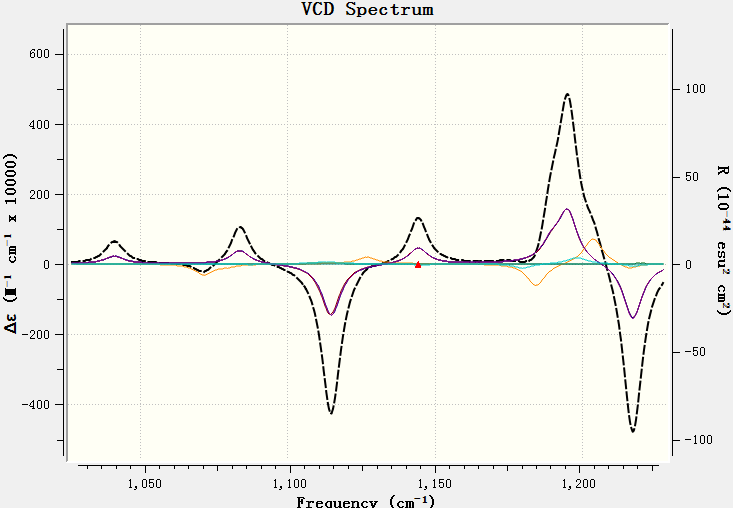
图18. VCD图谱:其中虚线为7个构象的Boltzmann加权平均图谱
图谱可以用数据点的方式导出,可以方便在Excel之类的工具里重新绘制、比较(比如与实验值比较)等等。
四. 相关教程
- Vibtrational Circular Dichroism. Exploring Chemistry with Electronic Structure Methods. 3rd Edition. Page281-289
- CONFLEX教程|构象搜索
- Gaussian教程 | ECD计算
- GaussView 6自带教程
- Gaussian教程 | 计算分子的拉曼光学活性(ROA)
- Gaussian教程 | 计算分子的比旋光度
- Gaussian教程 | NMR图谱与耦合常数的计算
强烈推荐阅读,采购该书:http://store.molcalx.com.cn/index.php/product/expl-chem
本教程提供了CONFLEX的详细操作流程,CONFLEX的试用与下载:http://www.conflex.net
本教程提供了详细的ECD计算操作步骤
GaussView 6 | Help | Tutorial | GMMX Conformer Tutorial


