
摘要:本文介绍了拉曼光学活性(Raman Optical Activity,ROA)的相关概念,并以α-pinene分子为例,详细介绍了如何利用高斯程序(Gaussian 16)计算分子的拉曼光学活性,并通过与实验测量光谱进行对比以确定分子的绝对构型。
作者:陈宇
日期:2019-12-26
基本概念
拉曼活性(Raman Activity)
当入射光与分子的振动模式相互作用时,会引起光子的非弹性散射,使得出射光的频率相对于入射光发生变化,这也称为拉曼活性。基于这种散射形成的光谱也称为拉曼光谱。
拉曼光学活性(Raman Optical Activity)
当入射光是圆偏振光时,对于某些分子,其对左和右圆偏振光的散射强度是不同的,这两种强度的差被定义为拉曼光学活性。它能为具有拉曼活性的分子提供关于分子手性和结构方面的信息。
拉曼光学活性(ROA)的计算流程
Gaussian程序提供了关键字(连同选项):Polar=ROA(指示程序计算拉曼光学活性)完成特定入射波长下拉曼光学活性的计算。计算ROA的基本流程包括:
- 结构优化与频率计算
- ROA计算
- 结果分析
首先优化分子结构,同时计算频率,通保存检查档文件(也就是.chk文件)。
读取上一步的检查档文件中的结构和波函数分别作为初始结构和波函数初始猜测,用更大的基组进行拉曼光学活性计算。
上述我们提到,拉曼光学活性能够提供关于分子手性中心的信息,这使我们能够通过比较计算和实验测得的拉曼光学活性光谱判断分子的绝对构型。
需要注意的是:为了精确计算拉曼光学活性光谱需要提供带有弥散函数的基组。同时,对于溶剂中测量的结果,需要考虑溶剂的影响。
算例演示
在本文中,我们以化合物α-pinene为算例进行讲解。α-pinene有两个手性中心(1R,5R),其对应异构体手性中心为(1S,5S),我们通过计算这个手性分子两个对映异构体的拉曼光学活性,并与实验测量的光谱进行对比,来说明如何利用这种手段鉴定手性分子的绝对构型。我们选用气相条件下测得的实验数据。
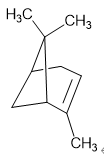
Figure 1. (1R,5R)-α-pinene的化学结构
第一步:优化与频率计算
优化α-pinene分子及其对映体的结构。
α-pinene结构优化输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 | %nprocshared=8 %chk=RR-a-pinene.chk %mem=1GB # apfd/6-311+g(d,p) opt freq RR-a-pinene 0 1 C -3.12040198 -1.16900277 -0.38642488 C -1.69901680 -1.09680250 -0.17875583 C -1.20674670 0.36753443 -0.19356806 C -1.75998369 1.20378109 0.68481009 C -3.05432408 0.72490548 1.31855995 C -3.19091075 -0.78595540 1.03713927 H -1.04263405 -1.64377509 -0.82286968 H -3.60886089 -0.59442120 -1.14547950 H -3.44347946 -2.17710510 -0.54215104 H -1.31729103 2.14591444 0.93241454 H -3.03706942 0.92802561 2.36896201 H -3.88951117 1.22609955 0.87565249 H -4.09513480 -1.09077317 1.52125186 C -1.84734481 -1.60431301 1.38172391 C -0.44194084 -0.99827125 1.55246661 H -0.52887947 0.02540761 1.85150384 H 0.09339291 -1.54425738 2.30094302 H 0.08614602 -1.05488302 0.62358590 C -1.93087711 -3.10301194 1.03745094 H -1.43037382 -3.67109051 1.79354829 H -2.95736852 -3.40100619 0.98831025 H -1.46292767 -3.27824293 0.09129164 C -0.10492662 0.83393865 -1.16313477 H -0.50569391 1.56570369 -1.83306426 H 0.70150741 1.26369718 -0.60647352 H 0.25495664 -0.00352503 -1.72352628 !空白行都是特意留着,不可省略 |
α-pinene对映体结构优化输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 | %nprocshared=8 %chk=RR-a-pinene-enan.chk %mem=1GB # apfd/6-311+g(d,p) opt freq RR-a-pinene-enan 0 1 C -3.12040198 -1.16900277 0.38642488 C -1.69901680 -1.09680250 0.17875583 C -1.20674670 0.36753443 0.19356806 C -1.75998369 1.20378109 -0.68481009 C -3.05432408 0.72490548 -1.31855995 C -3.19091075 -0.78595540 -1.03713927 H -1.04263405 -1.64377509 0.82286968 H -3.60886089 -0.59442120 1.14547950 H -3.44347946 -2.17710510 0.54215104 H -1.31729103 2.14591444 -0.93241454 H -3.03706942 0.92802561 -2.36896201 H -3.88951117 1.22609955 -0.87565249 H -4.09513480 -1.09077317 -1.52125186 C -1.84734481 -1.60431301 -1.38172391 C -0.44194084 -0.99827125 -1.55246661 H -0.52887947 0.02540761 -1.85150384 H 0.09339291 -1.54425738 -2.30094302 H 0.08614602 -1.05488302 -0.62358590 C -1.93087711 -3.10301194 -1.03745094 H -1.43037382 -3.67109051 -1.79354829 H -2.95736852 -3.40100619 -0.98831025 H -1.46292767 -3.27824293 -0.09129164 C -0.10492662 0.83393865 1.16313477 H -0.50569391 1.56570369 1.83306426 H 0.70150741 1.26369718 0.60647352 H 0.25495664 -0.00352503 1.72352628 !空白行都是特意留着,不可省略 |
第二步:ROA计算
使用更大的基组,从第一步结构优化保留的检查档文件中读取初始结构和波函数,并计算两种构型的拉曼光学活性。光谱的计算在气相条件,532nm入射光下执行。
(1R,5R)异构体的输入文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 | %nprocshared=8 %chk=RR-a-pinene.chk %mem=1GB # apfd/spAug-cc-pVDZ guess=read geom=check Polar=ROA RR-a-pinene-ROA 0 1 532nm !空白行都是特意留着,不可省略 |
(1S,5S)异构体的输入文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 | %nprocshared=8 %chk=RR-a-pinene-enan.chk %mem=1GB # apfd/spAug-cc-pVDZ guess=read geom=check Polar=ROA RR-a-pinene-enan-ROA 0 1 532nm !空白行都是特意留着,不可省略 |
第三步:结果分析
利用GaussView查看分子的拉曼光学活性光谱,方法如下:
- GaussView 6| File |Open 用GaussView读入ROA计算结果文件
- GaussView 6| Results | Vibrations
- 图谱绘制
- 将异构体的图谱展示在同一个图谱中
- 展示ROA图谱
- 图谱风格的设置
- 最终图谱的生成
首先,打开GaussView6,读入RR-a-pinene-ROA.log与RR-a-pinene-enan-ROA.log,使两个分子显示在同一个主界面(图2)。可以分别用鼠标将两个文件分别拖入到同一个主界面。
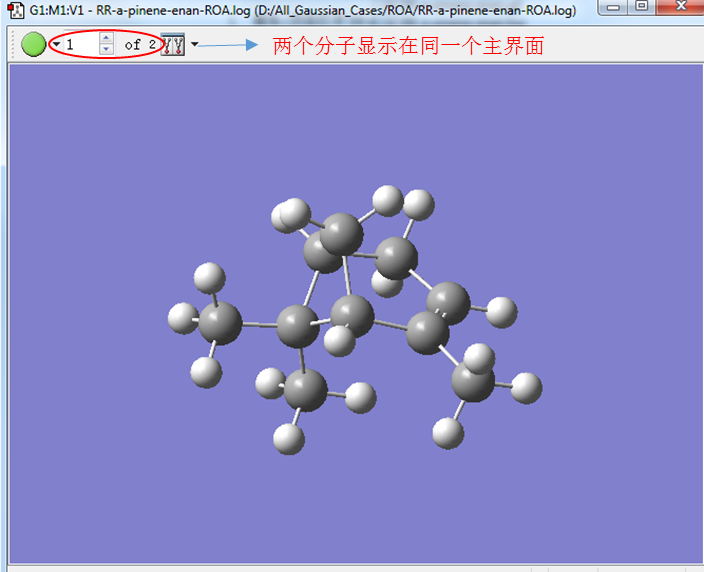
Figure 2. 用GaussView读入ROA计算结果文件
在Results下拉菜单中点击Vibrations选项,将显示不同频率下振动强度,拉曼散射强度等数值(图3)。
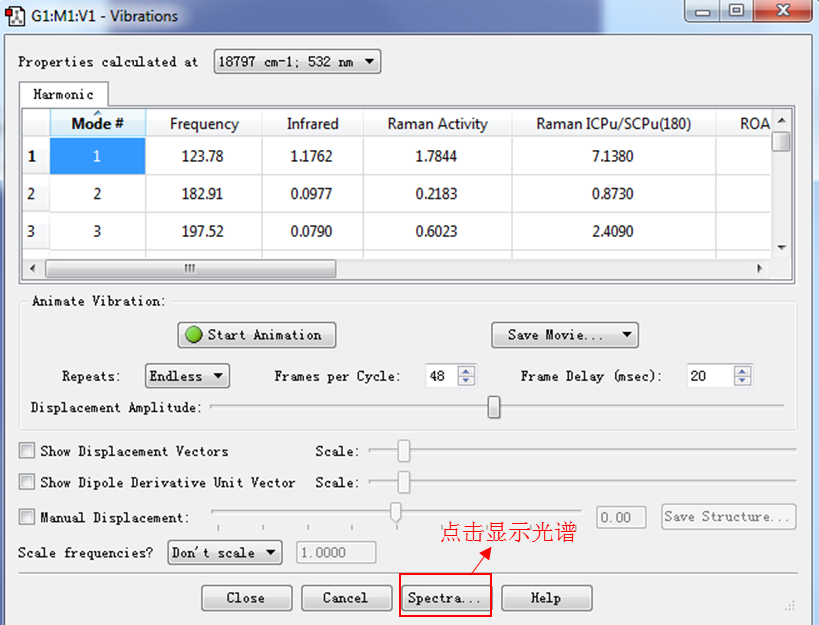
Figure 3. 打开GaussView的光谱绘图
点击上图对话框下方的Spectra选项,将显示红外,拉曼,拉曼光学活性等一系列光谱图(图4)。
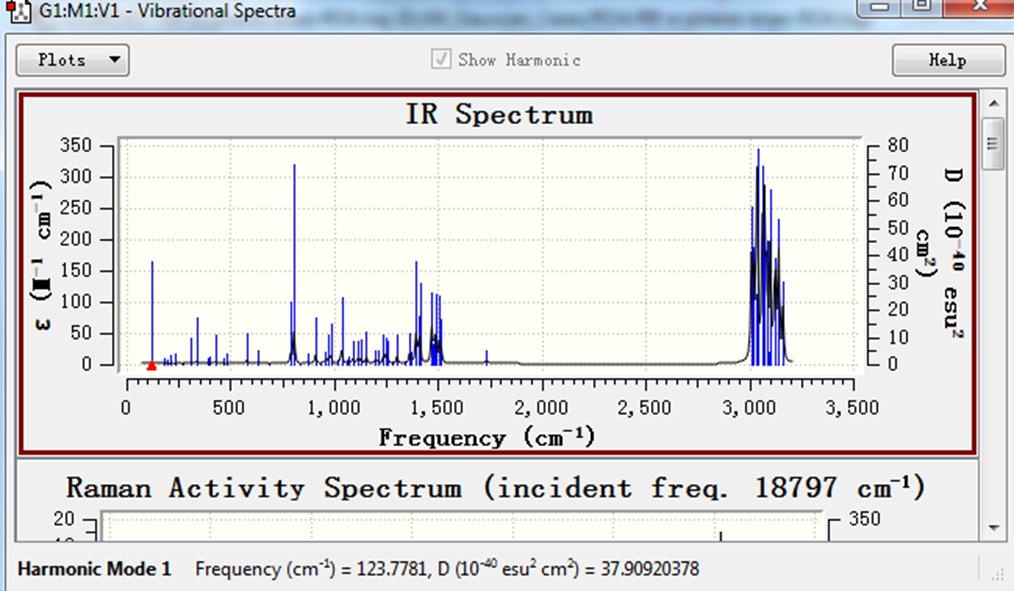
Figure 4. 图谱绘制
为了方便比较两个分子的光谱,我们可以点击Plots菜单下的Mixture Spectra选项将两个分子的光谱合并在同一个谱图中。
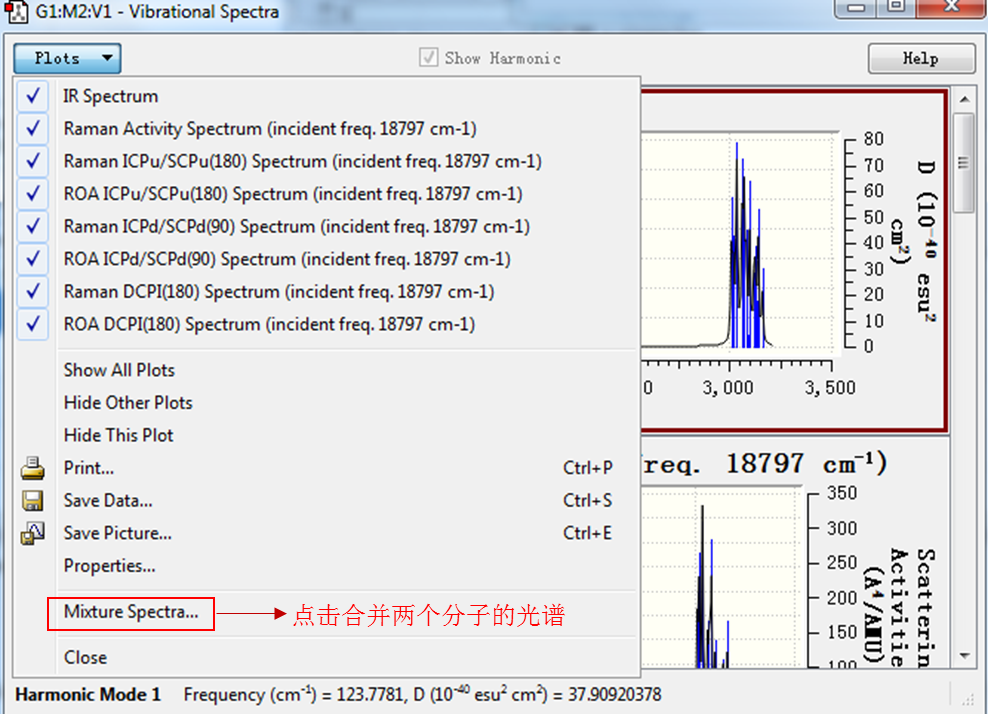
Figure 5. 将异构体的图谱展示在同一个图谱中
接下来为了更方便的显示我们感兴趣的拉曼光学活性,我们可以通过只勾选下面红框中的选项,从而只显示拉曼光学活性。
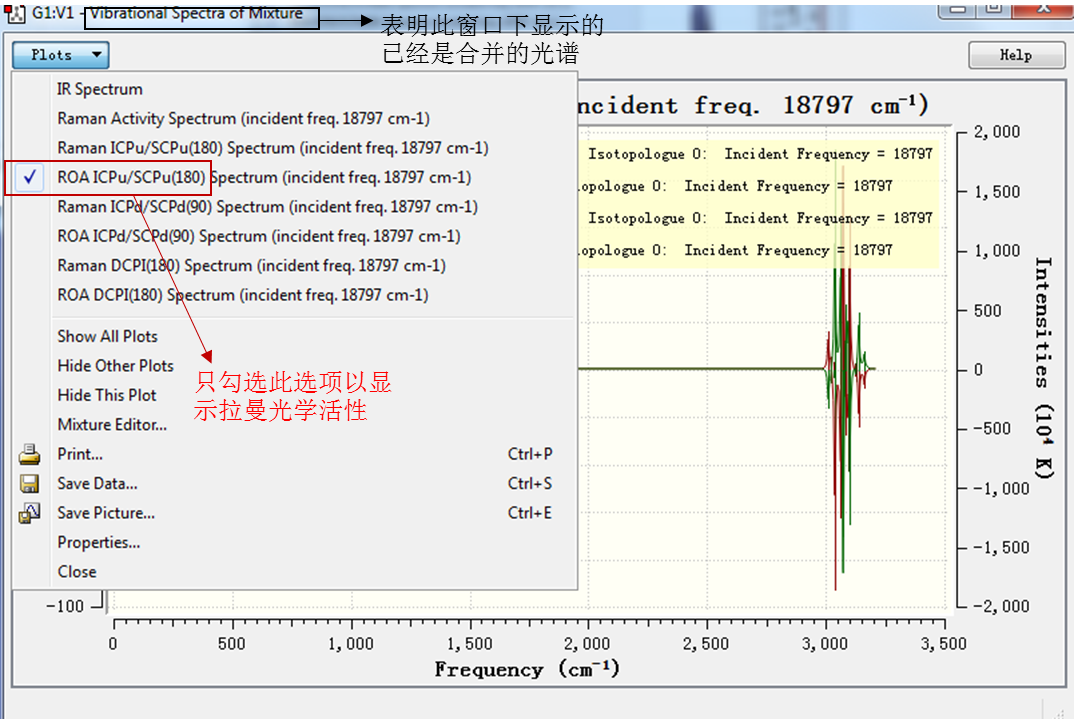
Figure 6. 仅展示感兴趣的图谱
需要注意:为了与实验测量光谱对比,我们选用标记为ROA ICPu/SCPu的选项。
为了更清晰的与实验谱图进行比较,通常我们会对光谱的显示进行一些处理,,首先,点击Plots菜单下的Properties选项,调出编辑对话框;然后勾选Invert Axis(使横坐标频率的数值从左到右递减,实验谱图通常如此)和Fixed Range选项,并在Fixed Range后面填入适当的值(依据实验测得的频率范围),最后点击Ok选项。
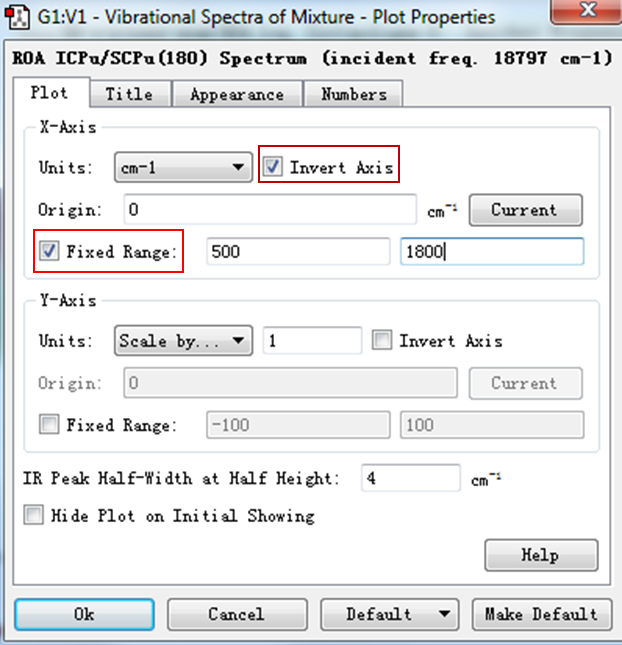
Figure 7. 设置波长范围
下图为处理好的谱图,可以看出只显示了1600-600cm-1范围的拉曼光学活性谱图。作为例子,我们在图中用黑圈标出了1500cm-1附近的三个特征峰值,与实验谱图(图8,我们也用红圈标出了三个相应的特征峰)对比,可以发现计算谱图中绿色线代表的光谱与实验曲线很好的吻合,这也说明实验中所用的分子构型为a-pinene(1R,5R),这也与其他手段得到的结果一致。
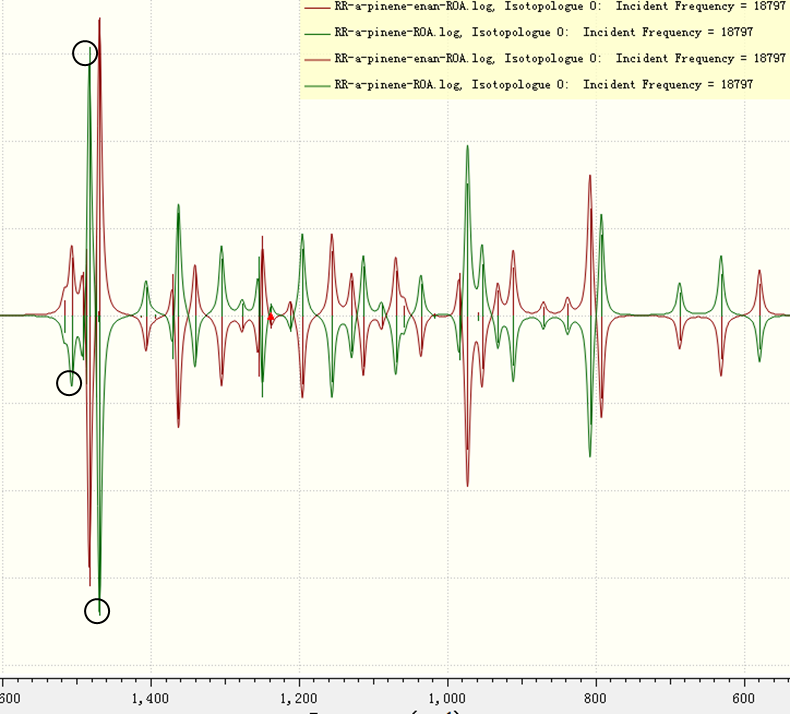
Figure 8. 1600-600cm-1范围的拉曼光学活性谱图
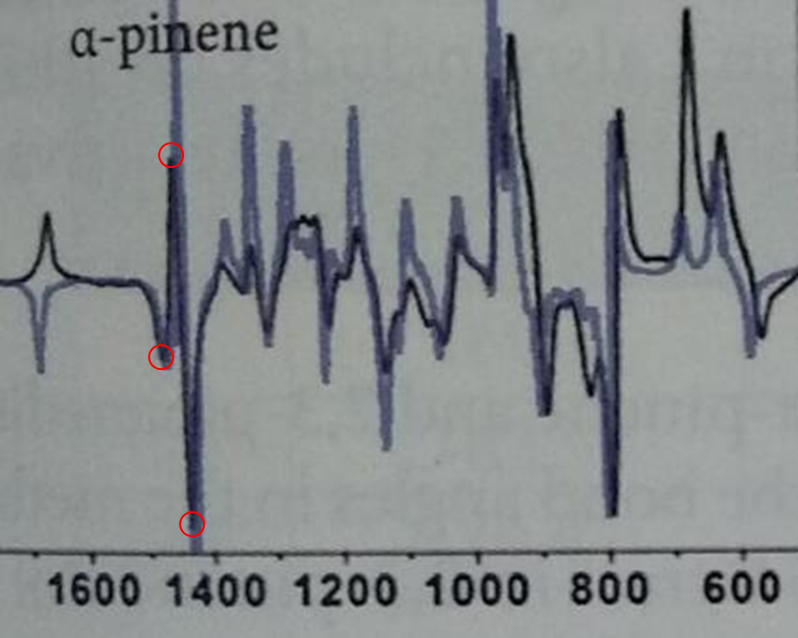
Figure 9. α-pinene的实验图谱
注意事项
对于柔性分子,需要进行构象搜索,对计算结果进行玻尔兹曼平均,具体方法参见NMR,VCD和ECD计算教程。
相关主题
参考文献
- Exploring Chemistry with Electronic Structure Methods, 3rd ed., Gaussian, Inc.: Wallingford, CT, 2015. J. B. Foresman and Æ Frisch


