
摘要:12个蛋白-配体体系的分子动力学模拟分别用于生成单一、基于结构的药效团模型,这些合并的模型联合使用了初始实验结构的信息与模拟过程中快照的信息。我们将合并的模型与对应的PDB药效团模型(即手工从实验结构生成的静态模型)进行比较,分析了每个药效团模型特征的频次、特征类型、以及没有出现在实验结构获得的静态模型中的特征。我们不仅观察到传统方法不能看到的药效团特征,而且还看到在分子动力学模拟过程中快速消失的特征以及由初始PDB结构得到药效团模型中可能是人为产生的特征。我们的方法有助于减轻基于结构药效团模型对实验结构单一坐标的敏感性。此外,在分子动力学模拟中出现的特定特征频次的分析有助于对每个特征进行重要性排序。
Wieder, M.; Perricone, U.; Boresch, S.; Seidel, T.; Langer, T. Evaluating the Stability of Pharmacophore Features Using Molecular Dynamics Simulations. Biochem. Biophys. Res. Commun. 2016, 470(3):685–689. https://doi.org/10.1016/j.bbrc.2016.01.081.
前言
药效团模型被定义为确保与特定靶标结构的最佳超分子相互作用(supramolecular interaction)并触发或阻断其生物响应所必需的立体和电子特征的集合[1]。这些特征包括氢键受体(HBA)、氢键供体(HBD)、正电与负电离子化基团(PI/NI),以及疏水区(H)与芳香环(AR)。基于结构的药效团模型使用蛋白质-配体复合物的3D结构来提取最佳相互作用和高生物活性概率所需基本的空间和电子功能决定点,目的是识别新的化合物。有多种不同的基于结构的药效团模拟软件,比如Schrodinger的E-Pharmacophores[3]、FLAP[4]与Ligandscout[5]。已经证明,在计算药物发现与优化中,从配体-蛋白复合物结构中获得基于结构的药效团模型是非常有用的[6]。
在使用基于结构的药效团模型时,我们会面临两个问题:模型对蛋白-配体复合物的坐标非常敏感;药效团模型的特征数量太少或太多[2]。第一个问题与蛋白-配体复合物坐标的来源有关。通常情况下,坐标取自于PDB数据库[7]。由于PDB中超过80%的蛋白-配体复合物结构是使用X-射线晶体学确定的,因此与晶体学有关的问题也适用于基于结构的药效团模型的构建[8]。特别是,通过X-射线晶体学解析的蛋白结构的细节可能会受到晶体接触和溶剂效应的影响[9];此外,人们对蛋白质-配体复合物中配体坐标的保真度提出了质疑[10]。蛋白和小分子天生就是动态的,表现为各种运动形式:从单个键的振动到整体、大型的结构运动。蛋白-配体复合物的晶体结构仅代表动态系统的单个快照,既不提供有关配体构象柔性的信息,也不提供有关结合口袋内/附近残基的运动信息[11,12]。因此,从这些结构衍生的药效团模型可能包含了由晶体环境或结构的单一坐标引起的非自然存在的特征。
此外,特征数太少(小于3)或太多(大于7)的药效团模型使用受限。 药效团特征数量的增加通常伴随着灵敏度的下降,通常需要选择数量有限的特征。 在这方面的主要问题不是识别到太多的特征,而是缺少确定其优先级的标准。参考文献[13]是一篇优秀的综述,可以从中找到有关的可能解决方案 [13]。
近年来,人们提出了几种在药效团模型中整合蛋白和/或配体分子动力学模拟的方法。多个复合物药效团模型来源于同一蛋白质与不同小分子接触时的多重晶体结构。从结构中提取蛋白-配体相互作用模式,并将其合并为一个药效团[14]。然而,这种方法仅限于具有多个晶体结构且具有同样结合模式的蛋白。
一种非常常用的避免依赖单一坐标的方法是利用分子动力学模拟(Molecular dynamics simulation,MDS)生成多套坐标并将其为基础生成药效团模型。人们证明MDS对于理解生物分子的动态性质[15]与溶剂效应[16]等是非常有价值的,也是诸如蛋白-配体结合自由能计算等等之类高级技术的基础[17]。有几个课题组用MDS生成蛋白-配体复合物的轨迹,然后对其聚类获得基于RMDS的结构以从中提取代表性的结构,然后对每个代表性结构生成药效团模型(见文献18的例子)。最近,Choudhury等人从MDS保存的每个结构生成药效团模型,然后用对接与筛选结果对其排序[19]。
在本研究中,我们采用与文献19相似的方式,对MDS保存的每个复合物结构计算了药效团。但是,我们用各个药效团模型得到一个一致的模型(Consensus model),该模型也整合了初始试验结构的药效团特征。本文,我们将这种药效团称为合并的药效团(merged pharmacophore model)其所有的药效团特征在实验结构与MDS生成的任意一个快照上均可看到。因此,该模型合并了蛋白-配体复合物的动力学信息。如果有需要,每个特征的频次可用来对特佂进行排序、检测离群的特征(即非常罕见的特征)。
我们研究了12个蛋白-配体复合物体系以测试我们的方法。每个体系在水溶液中进行了20ns的模拟,然后用前述的方式生成合并药效团模型。在本概念验证研究中,我们主要探讨三个关键问题:
- 传统的从PBD结构构建的药效团模型(PDB药效团)与合并的药效团模型之间有什么差异?
- MDS出现频次最高的特征是否也出现在PDB药效团模型中?
- 对每个复合物体系的研究除了回答上述两个问题之外,还探索了4种主要要笑谈特征(疏水、芳香、离子化基团与氢键)的行为特点。
除了将蛋白-配体复合物动力学信息合并于各个药效团模型之外,药效团特征出现频次的信息也有助于对特征进行排序,比如,出现于PDB药效团但在MDS中却很少出现(小于5%的时间)的特征可能意味着这不是自然出现的特征,可能需要放弃该特征。相反,没有出现在PDB结构但是非常高频出现在MDS(比如90%以上的时间)的特征,意味着该特征很重要。即使单独用频次信息不足以对特征进行排序,但是对应该选择哪些特征这样的决策提供了丰富的信息,尤其当药效团模型有很多特征的时候。
材料与方法
在本研究中,我们从PDB上选了12个不同的蛋白-配体复合物体系:1J4H, 1XL2, 2HZI, 3L3M, 1UYG, 3EL8, 2GTK, 2P54, 3BQD, 2AZR, 2OJ9与2OJG等等。这些复合物的选择相当随机,虽然采用了如下标准:体系的大小(溶剂化的蛋白-配体复合物小于70000个原子);仅有一个配体,结合时不涉及金属离子。复合物用其PDB代码来命名,并使用如下的术语。基于实验结构的药效团模型称为PDB药效团模型,PDB模型的药效团特征称为PDB特征。在合并药效模型中,所有的观察到的特征都映射到配体上,合并的模型包括了晶体结构的特征以及在MDS中出现的特征。没出现在晶体结构而仅出现于MDS的特征称为MD特征(MD derived feature)。
我们用CHARMM-GUI网页界面[20]对蛋白进行溶剂化处理、创建模拟作业。所有的MDS用CHARMM[12]执行。配体的参数与拓扑基于CGenFF Force Field[22]生成。蛋白-配体复合物用TIP3P水模型的立方体盒子进行溶剂化。每个体系的盒子的尺寸、总原子数、水分子数见补充信息的表1。在以1fs时间步长进行500ps的初始平衡后,每个体系在303.15K下进行了20ns的Langevin动力学模拟;蒙特卡罗气压调节器将压力保持在1个大气压左右。在生产计算(production calculation)时的时间步长为2fs;每10ps保存一次坐标,最后每个体系有2000个坐标。使用MDAnalysis软件包,通过计算回转半径以及蛋白和配体的均方根偏差来监视模拟的稳定性[23]。RMSD的计算方法如下:根据蛋白的Cα原子的坐标,将MD期间保存的所有坐标与起始结构进行拟合。将起始结构作为参照物,我们用重新定位的坐标计算了蛋白Cα原子的RMSD和配体重原子的RMSD。
在进一步分析药效团模型的时候,水分子不考虑在内。采用LigandScout 4.09.1为MDS保存的每一帧生成一个基于结构的药效团模型(2000个药效团模型),也为PBD结构生成基于结构的药效团模型[5]。
如手册[24]中所述,使用了默认特征约束来进行药效团模型的生成。每个蛋白-配体复合物有2001个药效基团模型,按如下方法进行分析。每个药效团特征具有两个属性:作为特征一部分的配体原子和特征类型。如果一个药效团特征的两个属性同时出现在两个模型里,则认为该特征是一样的。然后该特定特征的频数就增加一个。通过这种方式,我们获得了在MD模拟过程中特定特征出现频率的统计数据。针对PDB药效团模型中不存在的特征(即仅在MD模拟过程中看到的特征)进行了单独的统计。利用这一频次信息,通过将特征映射到配体的代表性2D和3D结构上,构建了包含模拟过程中看到的所有特征的合并药效团模型。
结果与讨论
所有的轨迹都经可视化检查以确保没有大的移动发生并且配体全程都呆在结合位点里。全部12个体系的Cα-原子RMSD都在可接受的范围内。大部分配体也是如此,除了2OJ9与2OJG之外。蛋白与配体的RMSD均值与标准偏差见补充材料的表2。在2OJ9算例里,配体的咪唑与吡啶片段在MDS期间可自由旋转,这使得配体平均RMSD为5.66Å。相似的,2OJG配体高度柔性的二甲氨基与苯基导致了高RMSD。但无论如何,这两个算例蛋白-配体复合物在模拟期间是稳定的。

图1. 合并的药效团模型映射到十二种蛋白-配体体系的配体二维结构上,各个图用PDB代码标记。特征类型的颜色编码如下:黄色球体表示疏水特征(H),灰色/白色国际象棋网格球体表示芳香环作用(AR),绿色小球体表示氢键供体(HBD),红色小球体表示氢键受体(HBA) ),蓝色小球表示正电中心(PI),粉红色大球表示负电荷中心(NI)。 方框中的数字为百分比,表示每个合并模型的药效团特征在各个单独的药效团模型中出现的频率(参见方法)。 方框颜色表示特征类型:H(黄色),AR(黑色),HBA(浅绿色),HBD(红色),PI(蓝色),NI(粉红色)。 填充框表示晶体结构中不存在的特征,即仅在MD模拟过程中出现的特征。
图1展示了全部12个体系的合并药效团模型。这些模型包含了所有遇到的特征、频次(百分数,图1小盒子里的数字)。因此,图1概述了12个复合物每个特征的稳定性/健壮性。
粗略地检查图1即可发现,所有蛋白-配体系统都有MD特征(即仅在MD模拟过程中出现的药效团特征),这可从列出频次信息方框的阴影背景可看到。但是,MD特征的数量变化很大,从1个(如2HZI)到5个(如2P54)。类似地,MD特征不但可以很少发生(少于5%的时间,例如1J4H),还可以非常频繁地发生(超过90%的时间,例如2GTK)。
图1所示的合并药效团模型大致可分为两类:(1)MD模拟补充很少的新信息的药效团模型,即任何MD特征均具有较低的频次,以及(2)MD模拟揭示了有助于进一步对模型开展工作信息的药效团模型,即模型的PDB特征在MD模拟里完全消失或仅很少出现,但MD特征则以较高的频率出现。
我们认为2HZI、1J4H、3L3M、2AZR和2p54是组(1)的成员。它们的大多数PDB特征的均显示为高频、MD特征显示为低频。尤其特别的是,2HZI和1J4H是第一类的原型成员:只观察到一个(2HZI)或两个(1J4H)MD特征,它们的频次非常低。将其它三个复合物指认为第1组有点模棱两可。例如,3L3M有8个MD特征,而且其中1个PDB特征在MD模拟中仅有17%的频次。然而,所有MD特征均很少观察到(1-8%),“低频”PDB特征比如芳香相互作用特征。如以下将讨论的,在MD模拟时,芳香特征通常倾向于以低频出现。因此,17%是芳香特征相对高的值。 正因为如此,我们认为最好将3L3M归属为类别(1);在2AZR和2p54的算例中,适用类似的论点。
我们认为2OJG、3BQD、EL8、1XL2、2OJ9、2GTK和1UYG属于第(2)类。它们都具有在MD模拟期间很少出现的PDB特性(1XL2,2OJ9)、具有高频的MD特性(2OJG,3BQD)或两者兼具(3EL8,1UYG,2GTK)。我们以2OJ9和2OJG为例来详细地说明。对于2OJ9的药效团模型,一些初始PDB特征在MD模拟期间是不稳定的。在MD模拟过程中,存在三个频率高于90%的疏水PDB特征,但两个PDB氢键特征的频率低于21%。在六个MD特征中,只有唯一一个氢键供体特征可在50%以上的时间内被看到。
2OJG的药效团模型仅有两个PDB特征:一个是疏水性,另一个是氢键供体。而合并的模型包含了三个新的MD特征(一个氢键供体,两个氢键受体), 三者的发生频率都很高(> 70%)。它们在PDB药效团模型中的缺失可能是由于蛋白结合口袋中配体的不利pose所致。
第2类的成员提供了说明动力学如何影响药效团假说的好例子。一方面,在MD模拟期间表现出低频的PDB特征可能是初始蛋白-配体坐标是不真实的。如果这些特征被保留用于虚拟筛选,则可能导致错误/误导性的苗头化合物。另一方面,MD特征对于构建有用的药效团模型是必不可少的。用2OJG的PDB药效团模型进行虚拟筛选是不可行的,因为为了获得有意义的结果至少需要三个特征。相比之下,具有五个特征的合并模型可作为虚拟筛选有用的起点。
图1说明了动力学对本文研究的12种复合物的各个药效团模型的影响,表1总结了MD模拟过程中特征类型的行为。对于疏水性(H)、氢键受体(HBA)、氢键供体(HBD)、负电中心(NI)、正电化中心(PI)和芳香环(AR)等六种基本药效团特征类型,表1列出了全部十二个合并模型中这些特征类型出现的累积次数(“merged features, total”列)。然后,该数字分解为PDB模型中存在的特征(“PDB features”列)和仅在MD期间可见的特征(“MD derived feature”列)。对于两类中每一类,给出了总出现次数,以及出现次数大于90%和大于50%模拟时间情况。
总的来说,PDB特征看起来比MD特征更稳定。仔细观察表1中的“Sum”行可以发现,79%(85个中有67个)的PDB特征可在MD模拟过程中有50%或以上的模拟时间出现,而只有24%(50个中有12个)的MD特征在MD过程中有50%以上模拟时间出现。如果考虑MD模拟期间出现频次>90%的总特征数,则这种区别变得更加明显:即PDB特征为66%,而MD特征为10%。
表1. 分子动力学模拟对不同类型特征的影响
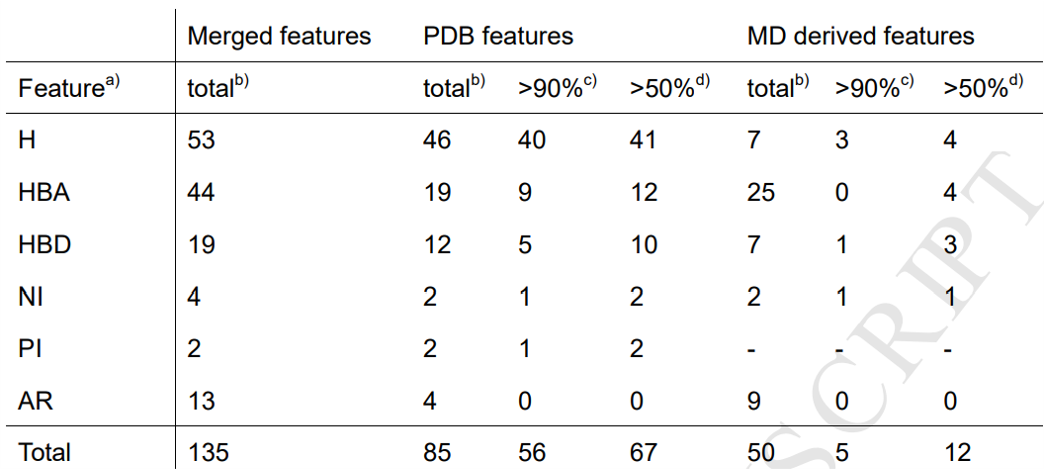
然而,同时表1说明了动力学对不同类型特征的影响相当不同。例如,芳香环特征(AR),MD的芳香环特征(9)明显多于PDB的芳香环特征(4)。这两种情况都很少发生,即对于AR,在大于50%和大于90%的列中所有的条目均为零。疏水特征(H)则发现了相反的情况。在此,PDB比MD的特征要多得多,特别是在PDB算例中,该特征的鲁棒性很高(43个疏水PDB特征中有40个具有90%的模拟时间)。
AR和H特征是最极端的示例,其他四种特征类型(HBA、HBD、NI、PI)位于这两种特征之间。 氢键受体和氢键供体类型在普遍性上位于疏水性特征之后。与疏水特征相似,PDB特征的稳定性高于MD特征。 70%以上的PDB氢键特征(HBA与HBD)属于>50%那一组;而MD氢键特征,这个数字下降到22%。然而,MD氢键特征(32)比PDB氢键特征(31)稍多。特别地,MD的HBA特征的数目(25)大于PDB的HBA特征的数目。
特征类型之间稳定性的差异与各种特征类型的标准/定义有很大关系。
疏水性特征的高稳定性是管理疏水性特征分类规则的结果。具体地说,在LigandScout中,大分子侧的相互作用伙伴可以位于1.0-5.9Å 之间的任何位置,并且配体和蛋白侧的原子基团被视为是疏水相互作用的一部分的要求是相当的非专一性[5,24]。类似的限制也适用于所研究体系中存在的少量电荷中心特征(四个NI,两个PI):配体中的可离子化基团被视为电荷中心特征(可离子化特征),LigandScout要求在1.5至5.6Å 范围内的大分子侧存在相反的可离子化基团。考虑到这些广泛的约束,这些特征在大多数的模拟时间内出现,前提是有一个相互作用的伙伴[24]。
相对大量的MD 氢键受体特征(HBA)是配体的化学性质(大多数配体具有可充当氢键受体的多个基团)和氢键相互作用定义的结果。LigandScout使用角度和长度标准对氢键特征进行分类,即相互作用的双方必须在指定的角度范围内,并且接近某个距离阈值[5,24,25]。一方面,与疏水特征相比,氢键特征的限制更为严格,几何结构的微小变化可以触发受体-供体对是否被归类为氢键。然而,同时,作为与氢键受体特征相互作用伙伴的氨基酸(苏氨酸、酪氨酸、赖氨酸、胱氨酸、谷氨酰胺、丝氨酸、组氨酸、精氨酸) 大多原子基团较小,在MD模拟过程中可以旋转和移动。因此,当特定的氢键断裂时,蛋白那边的伙伴通常容易被另一个氨基酸替换。因此,氢键特征的动力学行为不同于疏水特征的动力学行为(数据未显示)。
芳香特征的低频次是分类的规则导致的:芳香环平面上的几何约束很容易被柔性芳香环(例如可以旋转的环)触犯;此外,蛋白原子基团必须满足的规则是相当严格的,被视为对等的芳香相互作用[25]。
尽管在本研究中使用LigandScout生成药效基团模型,但基本的药效基团特征类型通常可用遵循相似规则和约束的软件解决方案中实现[2]。出于这个原因,使用其他药效团生成的软件可以得到相似的结果。
结果表明,MDS获得的频次信息可用于优化药效团模型,通过药效团特征增加、删减以及权重改变来实现。同时也清楚地表明,在不同类型的特征之间,其频次不可直接比较,至少不在一个线性尺度上。使用这些结果是第一步,本主题的工作会继续进行,并研究用MD模拟获得的信息来优化药效团模型的可能性。


