
摘要:3D药效团模型是生物活性构象的配体以化学方式定义的相互作用的三维集合。它以一种优雅的方式来解读化学编码的配体信息,因而在药物设计中成为一种有价值的工具。本文综述了3D药效团的基本概念及其在虚拟筛选和蛋白功能的机理研究中的应用。此外,还讨论了该领域的最新发展。将3D药效团模型与分子动力学模拟的结合使用是一个巨大的飞跃,因为该方法将大分子-配体相互作用视为动态的,因此显示出生理相关的相互作用模式。其它趋势包括在机器学习和人工智能应用中高效使用3D药效团信息或通过网络服务器进行3D药效团建模。最近的发展表明,3D药效团建模是一个充满活力的领域,在药物发现及其之后的研究中具有各种应用。
原文:Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T. N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next Generation 3D Pharmacophore Modeling. WIREs Comput. Mol. Sci. 2020, 10 (4):1–20. https://doi.org/10.1002/wcms.1468.
编译:肖高铿/2021-04-16
1. 前言
诸如蛋白或DNA之类的生物大分子与有机小分子的结合会触发其功能调节和生物反应。配体与其大分子靶标结合的方式是基于少量的化学相互作用(化学特征),如氢键、电荷或亲脂性接触等等。3D药效团是这些相互作用模式直观、有力的描述。它的高度抽象使化学结构多样的配体的结合模式获得合理解释,并进而实现了对分子数据库进行快速、高效地虚拟筛选。虽然3D药效团的概念是在19世纪初发展起来的,但直到80年代末和90年代初当第一批数据库搜索软件包发布时才能进行虚拟筛选实验[1]。分子量低于500Da的化学空间估计至少包含1060个有机分子[2]。此外,目前机器学习算法的发展允许以计算的方式生成数十亿个理论上可合成的分子[3]。3D药效团提供了一个独特的机会从巨大化学空间中获得类药分子。
2. 3D药效团的原理
3D药效团捕捉配体中与大分子靶标分子间相互作用相关的化学官能团的性质及其在空间中的三维排布。因此,化学官能团可按更常见的药效团特征进行分类,例如疏水性区域、芳香环系统、氢键受体、氢键供体、负离子化基团和正离子化基团[4]。有助于配体结合的不太常见的相互作用类型,例如金属配位和卤键,已经在大多数软件包中实现,或者通过用户自定义来实现[5-7]。除了化学特征和空间排布之外,3D药效团还捕捉氢键和芳香相互作用特征的方向[8]。此外,可以对每个药效团特征的空间容差(spatial tolerance)和权重进行微调,以调整其在3D药效团中的大小和重要性。为了描述结合位点分子的优选形状,药效团特征通常与排除体积限制(exclusion/excluded volume constraints)相结合。例如,排除体积限制可以由一组代表蛋白残基的球体组成,这些蛋白残基组成了结合配体潜在的边界。
表1. 3D药效团软件、组成及是否学术免费使用
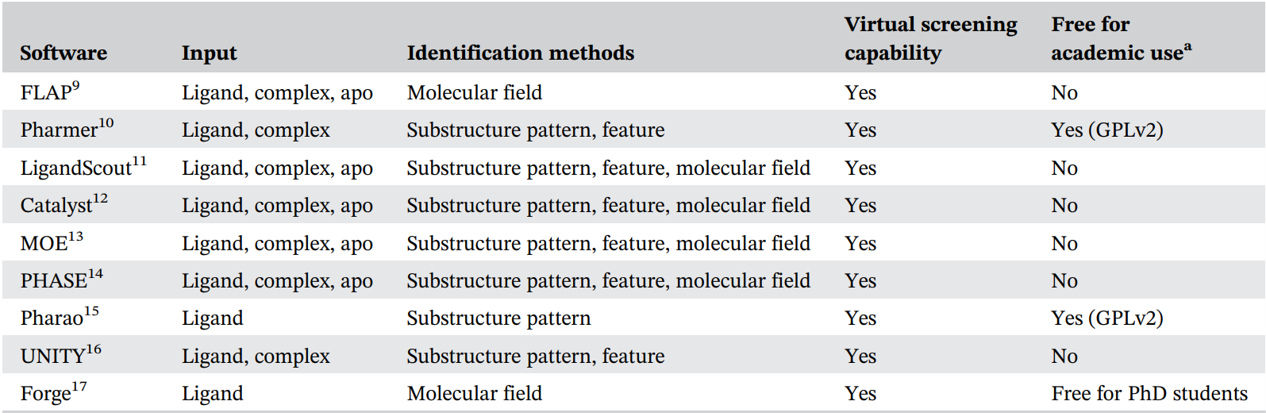
在已经开发的3D药效团建模程序中,有些对学术用户是免费的(表1)。尽管药效团特征及其确切定义和实现在不同的3D药效团建模程序之间可能不同,但是3D药效团的基本概念是一样的。
2.1 3D药效团的识别
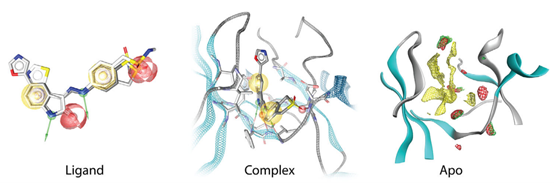
图1. 基于现有数据的三维药效团生成方法。3D药效团可以由一组已知配体(ligand)、配体-大分子靶标复合物的原子模型(complex)或单一的大分子靶标(Apo)结构产生。
3D药效团的识别方法可分为基于特征的、基于子结构模式的或基于分子场的,这取决于药效团特征是如何产生的。基于特征的方法通过过滤匹配分子相互作用特征的几何描述符来推导药效团特征。基于模式的方法(如PHASE、LigandScout和Catalyst中实现的方法)通过检测分子中的子结构来定义化学特征。例如,所有羟基被定义为氢键供体和受体。相比之下,基于分子场的方法(如FLAP和Forge)用不同的化学探针对配体或大分子靶标的分子表面进行采样,并计算相互作用能量图(energy map),这些能量图可以转化为药效团特征。3D药效团生成方法之间的另一个区别是所用数据的类型。这可以是一组活性配体、与大分子靶标复合物的配体结构生物学数据,也可以是单独大分子靶标的结构生物学数据(图1)。
2.1.1 基于配体的3D药效团
在缺乏大分子靶标的结构信息的时候,可以使用基于配体的3D药效团。它们由一组活性化合物共有的化学特征组成,这些化学特征对于化合物与靶标的相互作用很重要(图1)。公共的药效团特征通常源于活性化合物不同构象的3D叠合。将构象的3D结构进行叠合,以使得相同的药效团特征位于相似的位置。如果全部叠合的分子在特定位置共享某个特征,则将药效团特征放置在该位置[18]。3D叠合通常在基于快速距离检查的预过滤步骤之后进行,这会极大减少计算时间。例如,Catalyst中的HipHop算法使用“剪枝穷尽搜索(pruned exhaustive search)”法从分子构象中发现的双特征药效基团开始逐步建立共享3D药效团[12]。为了在每个步骤中识别出共享的3D药效团,需要一个预先计算好的分子特征间距离列表,首先对列表进行检查,以查看是否存在特定的特征组合。该预过滤步骤之后是对特征进行最小二乘拟合的叠合。LigandScout首先通过基于特征间距离来检查两组药效团特征之间的最佳配对,然后使用Kabsch算法进行叠合并确定最优叠合[19]。在某些软件包中,例如Catalyst中的HypoGen,推导出的化学特征三维排列与已知活性化合物的生物活性之间具有相关性[14,20,21]。此步骤可以帮助确定每个特征对于小分子生物活性的重要性。
然而,重要的是要注意到,分子的生物活性构象通常是未知的。因此,基于配体的3D药效团软件为每个分子考虑了一组低能构象。尽管商用构象生成算法通常能够成功重现生物活性构象,但基于配体的3D药效团生成过程不能保证能够用生物活性构象产生叠合结果[22]。基于配体的3D药效团的另一个局限性是对结构相似分子的依赖性,因为结构多样化的分子可能不共享相同的结合模式,并因此需要分别建立药效团模型。但是,即使不同的分子共享相同的结合模式,越是多样性的分子,基于配体的算法的正确叠合也就越有挑战性[8]。
2.1.2 基于结构的3D药效团
基于结构的3D药效团识别可以在两种类型结构的原子模型上进行。在大分子-配体复合物中,配体位于靶标分子的结合位点(图1)。配体主要通过共晶或对接到于靶标结合位点来得到与大分子的复合物结构。如果没有大分子-配体复合物结构可用,或着对于结合位点根本没有已知的配体,则程序可以从apo结构的原子模型中推导出3D药效团模型(见图1、表1)。 Apo结构是指没有配体结合的大分子的原子模型。
Apo 3D药效团识别技术在没有已知配体的情况下特别有用,这需要采用从头算法在结合位点里布置药效团特征。当然,apo 3D药效团生成方法也可用于大分子-配体复合物结构。在这种情况下,可为同一个活性位点生成全新的3D药效团,而不受现有配体的影响。这可用于在相同结合腔内探索新的化学空间区域。 因此,基于3D药效基团虚拟筛选的优势之一是通过不与任何特定配体结构结合的抽象特征的排布而提供的骨架跃迁(scaffold hopping)潜力。
即使蛋白是最常见的药物靶标,但是蛋白并不是3D药效团开发中分析的唯一大分子结构。包括LigandScout和Catalyst(表1)在内的程序可以基于核酸生成3D药效团模型。例如,Spitzer等人基于DNA-配体复合物结构的小沟结合位点生成了3D药效团模型[23]。
基于特征的方法可用于大分子-配体复合物以及空的结合位点。 基于特征的程序会分析靶标-配体复合物,并使用一套化学和几何规则来对靶标-配体的相互作用进行识别与分类,然后这些相互作用组成药效团特征[19]。在一个将基于特征的方法应用于apo蛋白结构的示例中, Schrödinger提出了一种策略,即使用Glide XP对接程序将片段对接至apo结合位点[24,25]。选择能量上最有利的片段对接pose,用“Phase”构建3D药效团模型(表1)[14]。
例如FLAP(表1)之类的基于分子场的方法利用分子相互作用场(MIF)来识别药效团特征放置的热点[9]。生成MIF的一个主要工具是GRID软件,该软件因其在抗病毒药物扎那米韦的发现中所起的作用而闻名[26,27]。原则上,在预定义的结合腔里放置均匀间隔的网格,并放置探针以对结合位点进行采样。这些探针采是各种分子片段,代表了最可能在大分子和配体官能团之间发生相互作用。下一步,计算探针和靶标结构原子之间的能量以确定相互作用位点。因此,这些探针可以识别出与大分子发生有利相互作用的位点。这些相互作用能产生MIF,MIF以能量等值图的方式描述靶标和给定探针之间的相互作用能如何随靶标表面而变化。基于分子场的程序将MIF能量局部最小值的点取出称为“热点(hotspot)”,并根据该点上能量最有利的相互作用的探针类型将其转换为药效团特征。还可以通过使用诸如AutoDock中的AutoGrid之类的非商业软件来进行基于分子场的热点检测,AutoGrid软件可获得各种原子类型的能量网格图[28,29]。
用于apo蛋白结合位点生成药效团特征的程序会产生多余的可能特征。 用于虚拟筛选,必须将特征数量减少到合理的水平:需要在足够多的特征以保证专一性与不要过多地使用特征以免太严格而导致假阴性之间取得平衡。 一些程序包含特征简化功能,但其它程序会输出初始的、未经优化的3D药效团。初始未经优化的3D药效团由许多特征组成,需要由用户对特征进行精简。可以根据结合位点和结合位内层原子的信息来选择特征,还可以根据用户希望配体利用结合位点的哪些特征进行选择。特征的精简不仅可以用手动执行的方式,如HS-Pharm[30]程序的一个示例,还可以使用机器学习来对初始的3D药效团特征数量进行精简,这将在后面的高级部分中进行讨论。
2.2 基于药效团的虚拟筛选
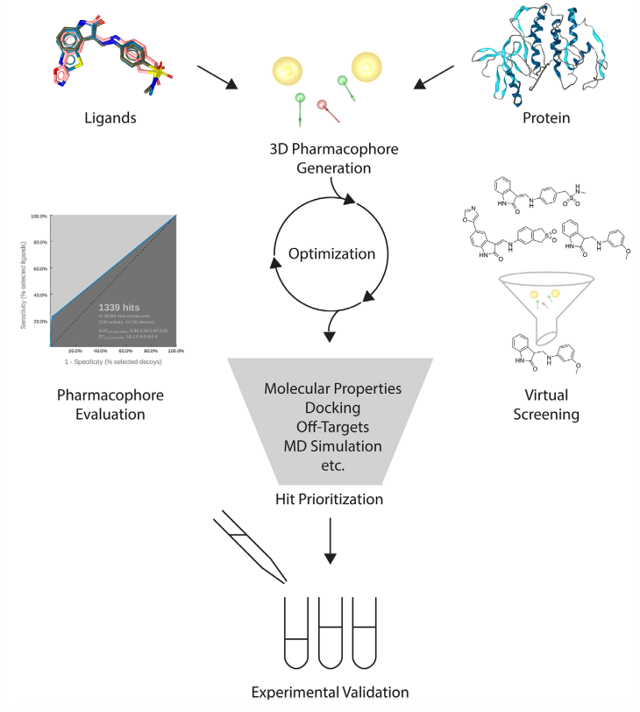
图2. 虚拟筛选工作流。3D药效团通过基于结构或基于配体的方法生成。最先进的回顾性验证是通过对活性(actives)与非活性(decoys)化合物绘制ROC曲线来进行的。在基于药效团的虚拟筛选之后,通常用诸如对接或分子动力学模拟之类计算昂贵的方法以进一步获取更多差异化的结构见解。ROC:receiver operating characteristics
在基于药效团的虚拟筛选中,3D药效团可从一组活性配体、靶标-配体复合物或apo靶标开发而得,并用于筛选分子虚拟库。将满足药效团query要求的分子共数据库中检索出来。与体外高通量筛选相比,虚拟筛选对化合物进行优先级排序可以显著提高命中率,从而减少进行实验测试化合物的数量(图2)。
为了解决分子的构象柔性,需要在筛选步骤之前准备好化合物的构象库。值得一提的是,不同筛选软件包处理构象的方式有所不同。一些软件包(例如LigandScout,Catalyst或MOE)对每个库分子的预先生成的一组构象用于虚拟筛选,而有些软件包(例如PHASE)以牺牲了虚拟筛选速度为代价可在筛选步骤时实时生成构象[19,31,32]。关于虚拟筛选构象生成的更多信息,请读者阅读相关主题的文献[8,22,32–36]。
在筛选步骤中,将药效团查询式(query)中的药效团特征与所筛选化合物库分子中的药效团特征进行比较。有两种不同的比较方法:基于指纹的方法和基于3D叠合的方法。诸如FLAP之类的基于指纹的方法主要将有关特征存在和/或特征几何形状的信息提取到类似指纹的描述符中,这使得能够在查询药效团和构象数据库之间进行节省时间的相似性比较(例如,使用Tanimoto系数)。诸如LigandScout、Catalyst和PHASE之类基于叠合的方法对药效团特征集进行3D叠合。如果分子不同构象的药效团特征集可以与查询药效团特征集叠合对齐,则报告匹配。3D叠合对齐在计算上既昂贵又耗时,特别是在大型分子库筛选的情况下。为了减少计算时间,通常在3D叠合对齐之前进行基于特征类型、特征计数或快速距离检查的快速预过滤步骤。LigandScout是唯一提供无损预过滤步骤的软件,提供几何上最精确的筛选算法。此外,其独特的模式匹配3D叠合对齐算法还可以筛选到其他依赖特征距离指纹算法的程序所不能发现的化合物[37]。
在结合配体的实验数据可用的情况下,可以对3D药效团模型进行验证。通常,3D药效团的验证集包含文献报告的活性(active)分子、非活性(inactive)分子和诱饵(decoy)分子。在准备验证集时,应考虑两点:首先,3D药效团描述了一个独特的结合模式(biding pose)。因此,活性化合物数据集的分子与靶标蛋白之间应该具有相同的结合模式。其次,应谨慎地使用文献报道的非活性分子,因为观察到的没有活性可能是由其他因素造成的,例如,在细胞活性测试中不能溶解或不能到达靶标。因此,我们鼓励使用精心挑选的诱饵(decoy)分子,而不是非活性分子。诱饵(decoy)是一种被认为是无活性的化合物,但是在物理化学性质上与活性化合物具有很高的相似性。DUD-E(Director of Useful Decoys)提供了一个便利网络工具用来生成诱饵分子[38]。对验证集筛选结果可用于评估开发的3D药效团模型的质量,并进一步优化它(图2)。
在用虚拟筛选的性能表现来评估3D药效团模型的质量时,可以使用各种指标或富集参数(图2)。3D药效团性能是根据3D药效团可以从数据集中搜索到多少个活性化合物以及3D药效团能够正确地将化合物分类为活性或非活性化合物来评估。富集参数将数据集中的化合物分为四类:活性(真阳性,TP);非活性但被识别为活性(假阳性,FP);非活性(真阴性,TN);活性但被识别为非活性(假阴性,FN)。不同的指标衡量3D药效团不同方面的性能。这些指标包括活性物质的产率(YA,yield of actives),它描述了3D药效图搜索到总命中数中存在的真阳性数[39]。ROC曲线绘制了真阳性比率与假阳性比率的关系,从而呈现了3D药效团模型的敏感性和特异性,表征了该3D药效团为了识别一定数量活性化合物时需要附带错误识别多少非活性化合物为活性化合物[39-41]。关于富集参数的综合列表,及其如何计算与用途,请参考读者Braga和Andrade等人的文献[40]。
在开发了3D药效基团并经回顾性验证之后,就可以用于筛选的商用或内部化合物库(图2)。 取决于3D药效团的复杂度以及化合物库的大小,药效团筛选会搜素到各种不同大小的命中列表。在三维药效团筛选之后,通常进一步用分子对接、分子动力学模拟或其他方法进一步表征结合模式,以获得更多的结构信息,从而理性对有待进行实验测试的分子进行优先级排序(图2)。
3. 应用案例
3.1 虚拟筛选
除了分子对接以外,3D药效团也广泛用于虚拟筛选。在本节中,我们重点介绍并讨论虚拟筛选的最新成功案例,涵盖了不同的靶标类别和方法。
3.1.1 用小分子调节剂平衡免疫系统
Toll样受体(TLR)在识别与感染和非生理性组织损伤相关的分子模式来激活先天性免疫反应中起关键作用[42]。小分子TLR调节剂的理性设计是治疗自身免疫性炎症、癌症、过敏或鉴定疫苗佐剂有前景的策略[43]。
2014年,Murgueitio和他的同事们面临着一个数据稀少的情况,没有可用的有机小分子TLR2抑制剂[44]。因此,他们基于MIF生成了一个3D药效团,以确定配体结合所需的关键相互作用。他们仔细开发了一个基于结构的药效团模型。随后的虚拟筛选发现了低微摩尔范围内的新型拮抗剂,20%的虚拟命中化合物具有生物活性。随着更多的有机小分子TLR2配体被报道出来,Murgueitio及其同事继续努力寻找新的TLR2调节剂,联合使用3D药效团与基于形状的虚拟筛选方法发现了新型的没食子酸(邻苯三酚)衍生物(MMG-11)[45,46]。
2019年, Šribar及其同事进行了基于结构的虚拟筛选,随后进行了体外实验验证,以寻找新型TLR8调节剂[47]。进行了分子对接以探索能够解释已知调节剂活性的不同结合模式。最具描述性的结合模式被转化为3D药效团模型,随后用于虚拟筛选。此方法最终发现了一种新型TLR8抑制剂,其中36%此虚拟筛选方法搜索到的分子具有体外活性。
3.1.2 新型共价结合配体的发现
共价结合配体的设计在药物发现社区越来越受欢迎[48,49]。延长的靶标停留时间和不受药代动力学影响的药效学效应允许更低的给药剂量或更长的给药间隔,使共价结合药物对许多疗法具有吸引力[50,51]。共价对接和量子力学(QM)计算代表了开发共价结合剂的“金标准”[52,53]。由于高昂的计算成本和时间,对接适用于筛选小型化合物库,而QM计算仅适用于单个化合物。对于大型数据库的虚拟筛选,基于药效团的方法更适用。Schulz及其同事在Ligandscout框架中引入一种称为“残基结合点”的新型药效团特征,该特征可以识别类药化合物的弹头(warheads),例如酮、腈或迈克尔受体[11,54]。
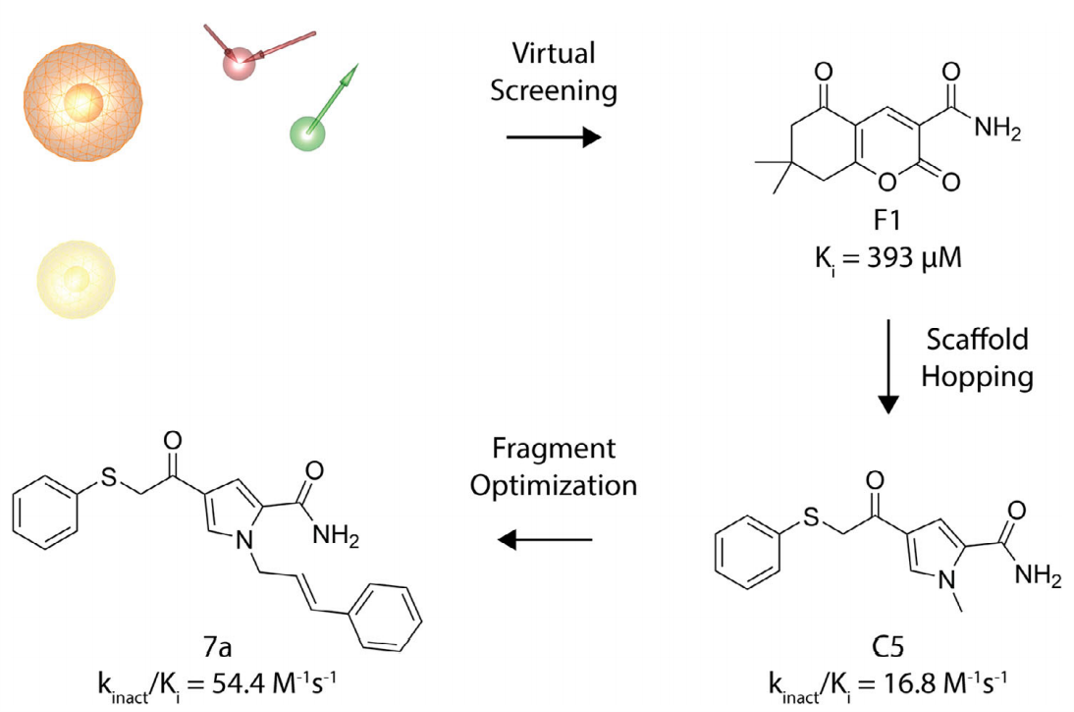
图3. 病毒3C蛋白酶共价抑制剂的发现。初始的片段是通过3D药效团筛选发现,然后经过骨架跃迁与片段生长进行优化。绿色箭头:氢键供体,红色箭头:氢键受体,黄色球体:疏水相互作用,橘黄色球体:残疾结合点(共价特征)
他们将残基结合点特征(也称为“共价特征”)用于病毒3C蛋白酶抑制剂的从头设计[54]。产生了一个选择性与特异性3D药效基团,其中包括对底物识别至关重要的非共价相互作用以及新的共价特征。将获得的3D药效团用于片段库的虚拟筛选。选择共价和非共价形式具有高度相似的对接结合模式的化合物用于体外测试。化合物F1,一种杂环芳族酮,在酶学测试中显示出最高的抑制活性(图3)。蛋白质谱法证明了该化合物可以与柯萨奇病毒(Coxsackievirus,CV)B3蛋白酶发生了共价结合。用骨架跳跃策略对化合物F1进行了优化,得到了更稳定、活性更强的苗头化合物C5,一种苯硫基甲基酮。用合成方法对该化合物进行修饰得到化合物7a,这是一种选择性且不可逆的CV B3和肠病毒(Enterovirus EV)D68蛋白酶的共价抑制剂。该示例说明了3D药效团模型不仅适合于识别新型配体,而且还适合于苗头化合物(hit)和先导化合物优化。此外,该研究还凸显出了3D药效团在越来越受欢迎的基于片段的药物发现中的适用性[55]。
3.1.3 用3D药效团靶向GPCR
鉴于蛋白偶联受体(GPCR)在人体组织中无处不在、药物可及、在生理和病理过程中起到调节作用,因而是非常重要的药物靶标[56]。也因此广泛被作为靶标而出现在寻找新型的生物活性配体的虚拟筛选中。
2017年,Frandsen及其同事使用内部开发的方法建立了一个组胺H3受体(H3R)药效团模型,用于虚拟筛选[57]。从GPCR晶体结构中提取配体-残基片段,并将其映射到靶标受体同样保守的结合口袋残基上[58]。该方法允许对结构数据不足或配体未知的孤儿受体进行基于结构的建模。由于其依赖于仅有的62个GPCR晶体结构中现有的配体-受体片段,最初的H3R药效团模型遗漏了一个重要的阳离子特征,后来通过对接研究将已知H3R配体拟合到apo药效团模型而得以增加。药效团特征使用Phase进行放置[14]。虚拟筛选、命中化合物的选择和有效配体的衍生化总计得到76个化合物,用IP1积累和放射性配体结合试验进行药理学测试。五个中性拮抗剂和一个反向激动剂具有低微摩尔水平的结合,命中率为8%。
随后,Schaller及其同事采用了另一种方法,使用配体引导的同源建模策略来发现新型的H3R配体[59]。该方法的要点是根据其解释9个抑制剂拮抗剂结合的能力来对1000个同源模型进行优先级排序。 随后,将所选的同源性模型用于LigandScout的3D药效团模型生成[11]。互补地,将10种不同的配体对接到H3R并用相互作用模式进行评估。用一组含有100个多样的活性化合物以及3051个诱饵分子(decoy)的数据集对药效团模型进行迭代优化与验证,最后选用三个性能最好的3D药效团模型用于并行虚拟筛选(parallel virtual screening, 小编注:这不是指并行计算,而是一种筛选策略,是指将三个模型同时用于虚拟筛选、并合并结果的虚拟筛选)。随后,选择了八个命中分子进行生物活性测试,其中两个化合物在放射性配体耗尽分析中表现出纳摩尔的亲和力,命中率为25%。
3.1.4 设计多靶标分子以控制炎症
花生四烯酸(AA)级联是炎症反应的关键生化通路,包括通过5-脂氧合酶激活(FLA)蛋白产生促炎脂质介质(如白三烯)与抗炎介质(如环氧二十碳三烯酸)。后者通过可溶性环氧化物水解酶(sEH)水解。因此,两种酶的同时抑制代表了控制源自AA炎症介质有前景的方法。 Schuster及其同事应用基于药效团的虚拟筛选发现了首个FLA/sHE双重抑制剂,其活性在纳摩尔水平[60]。第一步,他们用两种不同的基于配体的药效团模型对SPECS数据库进行了虚拟筛选,这两种模型都是从已知的FLA蛋白抑制剂中衍生出来的[20]。进一步用先前报道的基于结构的sHE药效团模型对上一步命中的化合物进行优先级排序,并选了20个用于活性测试,最后发现了一个新型、有效的FLA/sHE双重抑制剂[61]。由于多靶标方法越来越受到关注,本示例表明多靶标结合的配体发现可从基于配体的药物团中获益。此外,它还凸显了联合使用基于配体和基于结构的模型在多靶标药物发现中的应用潜力,其中一个3D药效团可作为另一个药效模型命中化合物列表的优先级排序工具。
3.1.5 在农作物科学中设计与优化新化合物
Yao等人通过靶向植物病原真菌中的酶,证明了基于配体的设计在作物科学中的成功应用[62]。该小组进行了基于药效团的虚拟筛选,以寻找琥珀酸脱氢酶(SDH)的抑制剂。 该蛋白与真核生物线粒体的电子传递链有关,是一种有效的杀虫剂靶标。所有市售的SDH抑制剂由偶联到芳香酰胺上的羧酸盐/酯和胺部分组成。Yao等人用Catalyst生成了基于配体的3D药效团模型,其包含了芳香、亲脂和酰胺等特征。 对三维药效团进行了验证并将用内部开发的酰胺集中库进行了虚拟筛选。为了构建酰胺集中库,使用Discovery Studio的Enumerate Library by Reaction protocol将多样的、有商品供应的羧酸酯片段连接到苯胺上,以探索羧酸酯母核的化学空间。对集中库进行虚拟筛选之后,选择了16个化合物用于体内测试。8个化合物在100mg/L浓度下对3种不同真菌的抑制率作用超过50%。对活性最高和最广谱的配体通过合成进一步优化。在第二个酰胺集中库中,将多样的胺连接到最佳的羧酸酯母核上,并用3D药效团进行筛选,所得衍生物进行SDH体外活性测试。通过联合使用计算与实验优化步骤,开发了一种广谱活性的、新型的、低摩尔活性级别的酰胺SDH抑制剂。
3.2 理解蛋白的功能
除了描述配体结合模式和虚拟筛选应用外,3D药效团是在机理水平上研究配体依赖性蛋白功能的强大工具。以下各节展示了3D药效团模型在理解药理作用机理中起关键作用的示例
3.2.1 模拟代谢
磺基转移酶(SULTs)在II相代谢中起着重要作用,由于其高度柔性与广泛的底物特异性而成为具有挑战的靶标。Rakers及其同事开发了基于药效团的SULT预测模型以区分底物、抑制剂以及依赖于浓度而同时即使底物也是抑制剂的配体[63]。这项研究之所以引人注目,有两个原因。 首先,它用分子动力学(MD)模拟得到酶的不同构象的系综来生成构象特异性的三维药效团模型。其次,将药效团拟合评分纳入基于支持向量机的机器学习方法中,对筛选结果进行后过滤。所得到的基于药效团的预测模型成功地应用于SULT1E1配体类型的筛选和分类,并增强了我们对SULT酶特异性的理解。
芳香烃受体(aryl hydrocarbon receptor,AHR)是一种配体依赖的转录因子,控制生理物质和异源物质的代谢。 基于经过仔细验证的同源模型,Tkachenko及其同事应用3D药效团模型来研究生理配体和外源性物质在AHR转运到细胞核和随后诱导CYP1A1方面的差异[64]。组氨酸291被认为是控制这两种功能的关键残基,但在与生理配体(如犬尿氨酸)和外源物质(如 β-萘醌)的结合中起到不同的作用。
3.2.2 研究配体依赖的蛋白功能
GPCR代表着一类具有高度复杂药理学以及以配体依赖方式调节受体功能的多种可能性的重要药物靶标类别。该领域的一个主要问题是受体选择性,特别是同一家族密切相关的亚型。为了了解毒蕈碱受体(muscarinic receptor)的二合一配体(bitopic/dualsteric ligand, 编译注:bitopic ligand是指能同时与正构及别构位点结合的配体)的亚型选择性,Bermudez及其同事使用3D药效团模型来识别亚型专一的相互作用模式[65]。一方面,理性解释了配体如何对特定受体亚型实现选择性;另一方面,确定了细胞外环区中的关键残基(例如M3特异性盐桥),这些残基负责亚型特异性受体功能。 M2受体的某些上述双结合位点配体表现出一些出乎意料但又令人感兴趣的药理特性,例如部分激动和通路特异性受体激活(信号传导偏好)。为了理解这些效应,将3D药效团模型与其它建模技术和药理实验相结合进行研究[66,67]。部分激动作用可以通过多种结合模式的存在来解释,这些结合模式稳定了受体的不同激活状态。该概念已通过实验验证,并通过理性设计得到完全激动剂,其只能采用稳定活性受体状态的结合模式[66]。以相似的方式研究了配体偏好的通路特异性,并建立了机理模型,提出了细胞外环区构象限制的关键概念残基[67-69]。在另一项研究中,在毒蕈碱激动剂中研究了光开关偶氮苯母核的氟化效应。这项研究表明,由于存在其他相互作用的可能,光开关的氟化不仅改变了光致变色行为,而且还改变了M1受体的药理学特性[70]。在所有这些GPCR研究中,3D药效团不仅是理解配体依赖性受体功能的关键,而且事实证明它还是合成化学家和药理学家进行交流的最佳工具。
4. 应用3D药效团原理的高级方法
在前面的章节中,我们概述了3D药效团的概念以及可使用的成熟软件。此外,我们介绍了几个使用3D药效团进行前瞻性虚拟筛选以及理解蛋白功能的最新应用案例。但是,什么可以被视为是一种先级的方法呢?通常,3D药效团是从大分子的原子模型或从多个配体构象的叠合而产生的,并用于分析结构-活性关系或进行虚拟筛选。此外,进行虚拟筛选实验通常需要进行本地的软件安装以及高性能计算机的使用。在以下章节中,我们将介绍集成MD模拟的构象、采用机器学习算法、提供3D药效团搜索服务而无需昂贵的授权费和高性能计算等等高级方法(表2)。
表2. 应用药效团概念的高级方法
4.1 整合分子动力学模拟的信息
由于大分子和配体都是动态的实体,因此很明显,大分子-配体配合物及其潜在的相互作用也是如此。根据这个概念,Carlson及其同事整合了MD模拟中的信息,开发了一个增强的3D药效团模型用于虚拟筛选新型HIV-1整合酶抑制剂[92]。随后,Carlson用HIV-1蛋白酶实验证明了,从 28个NMR构象系综生成的3D药效团模型要比从90个X-射线衍射结构生成的3D药效团模型的性能更好[93]。这项开创性的工作启发了其他研究人员,并开始了开发采用MD模拟的构象来生成3D药效团的方法。
4.1.1 水合位点约束的药效团(Hydration-site-restricted pharmacophore, 2012)
从apo蛋白结合腔生成、未经优化的3D药效团模型通常包含太多的药效团特征而无法进行有效的虚拟筛选。水合位点约束的药效团(HSRP)方法旨在通过识别蛋白表面的水合位点来减少药效团特征的数量,MD模拟计算可以得出蛋白表面具有不利热力学性质的水分子[71]。这种约束的3D药效团同时也更容易搜索到熵有利的配体。已用三种药物靶蛋白对HSRP方法进行了评估,成功地减少了药效团特征空间并降低了算力要求。
4.1.2 SILCS-Pharm(2014)
SILCS-Pharm利用MD模拟得到的探针分子结合热点(hotspot)来生成3D药效团[72,73]。配体竞争饱和位点识别(site identification by ligand competitive saturation,SILCS)方法用MD模拟对蛋白表面进行采样,用不同探针分子(比如苯环的碳作为芳香作用特征)重新装配出药效团特征性质。得到不同探针分子的概率图(FragMap)用玻尔兹曼转换法表示为自由能。这些自由能FragMaps最终被转换为药效团特征,并且相关的自由能在3D药效基团模型生成中用于特征选择的优先性排序。作者认为,用SILCS-Pharm法生成的3D药效团通常比各种对接方法以及上述HSRP方法生成的3D药效团性能更佳。SILCS-Pharm已用于指导靶向癌蛋白Mcl-1和Bcl-xL新型抑制剂的结合模式预测[95]。
4.1.3 Dynophores (dynamic pharmacophores,2015)
与基于MD从不同蛋白构象系综中收集药效团信息的方法相反,Dynophore实现了从基于MD构象采样中全自动地识别基于化学特征的相互作用模式[66,74]。动态药效团(dynamic pharmacophore,Dynophore)根据所涉及的配体原子及其特征类型,从轨迹的每一帧中顺序提取相互作用点(如氢键、电荷或亲脂性接触)。通过对发生频率以及与蛋白的相互作用模式进行统计表征得到超特征(super-feature)。 三维体积特征密度云提供了关于相互作用的空间分布信息,条形码图以时间分辨的方式显示特征的出现。 ilib/LigandScout内置了一个Dynophore应用程序,解决了经典3D药效团模型的两个缺点:静态特性和特征的几何简化。
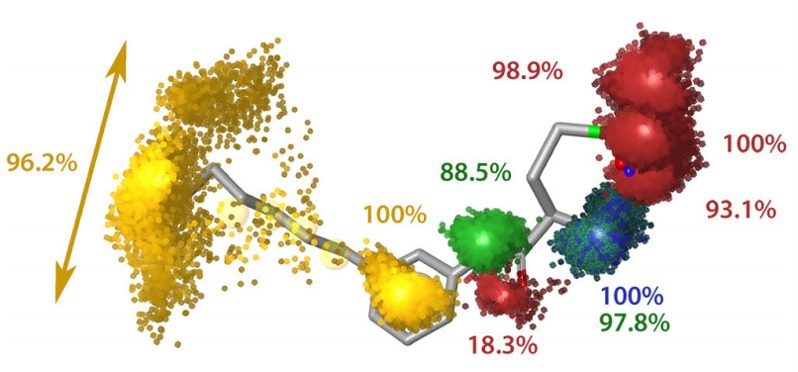
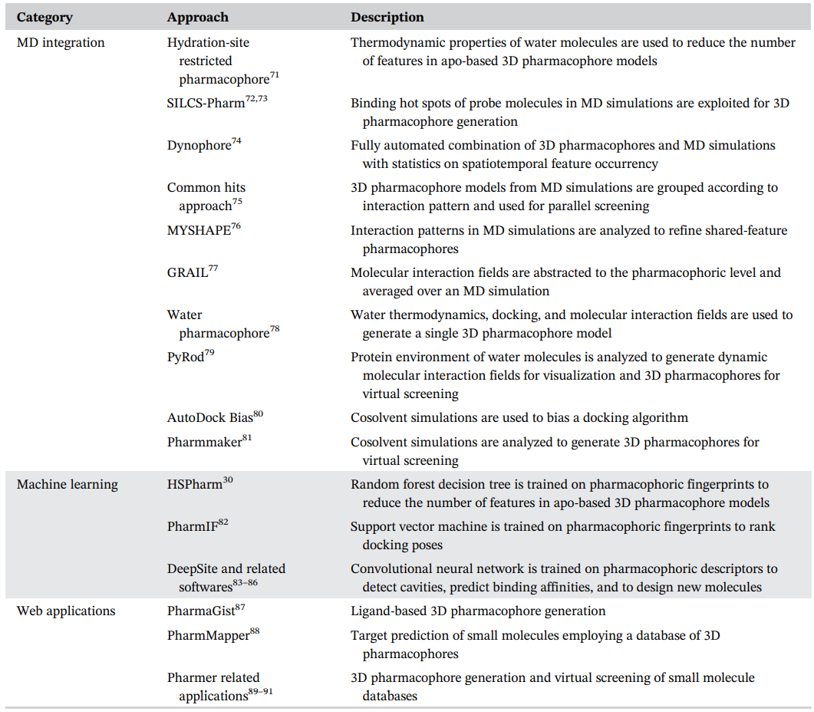
图4. Dynopores(dynamic pharmacophores)揭示了在100ns MD模拟过程中鞘氨醇-1-磷酸受体的配体ML056的动态结合模式变化。黄色点云表示疏水性相互作用,红色和绿色特征分别表示氢键受体与供体,带正电荷的区域显示为蓝色点云。特征旁边的百分比指的是它们在模拟期间的出现频率。在本算例中,分子的大部分保持在其初始方向上,从而导致相应特征点云的几乎为球形分布(右半部分)。 亲脂尾在结合位点内更加柔活,呈现出香蕉状特征云(左半部分)。
有些研究证明了dynophore是必要的,可以合理地解释单独采用经典3D药效团无法揭示的现象。Dynophore首先用于解释M2受体上两个配体的活性悬崖,它们的结构仅存在二氢异恶唑与异恶唑片段上单、双键的差异却具有相同的静态3D药效团[66]。基于发生频率和氢键受体的各自几何特性,两个配体之间不同强度的氢键作用可以合理的得到解释。在另一个例子中,Dynophore能够揭示能够揭示克服HIV-1逆转录酶(RT)耐药性机制。在该研究中,RT抑制剂rilpivirine通过与其他残基相互作用来绕过耐药突变,从而稳定了结合口袋中的抑制剂[96]。在针对金属酶精氨酸酶的虚拟筛选中,采用Dynophores来探索小分子抑制剂存在时结合口袋的可塑性,并提出了增加亲脂性相互作用的可能性。这样就产生了两种新型的片段样精氨酸酶抑制剂,有助于抗癌药物的开发[97]。Dynophore方法使研究人员能够摆脱经典3D药效团方法的静态性质,并为将结合模式作为动态事件进行描述和分析提供了新的机会(图4)[47,66,96-101]。表征超特征(super-feature)的特征分布概率密度函数可以直接用于虚拟筛选,为高效地将分子动力学模拟信息整合到快速有效的虚拟筛选中提供了新的可能性。
4.1.4 Common hits approach (CHA)和MYSHAPE(2017)
这两种方法基于MD模拟选择和优化3D药效团。 CHA根据相互作用模式从MD模拟的蛋白-配体构象里获得3D药效团模型,并对3D药效团模型进行分组得到代表性的3D药效团,随后用于计算成本高昂的虚拟筛选[75]。用40个蛋白-配体的体系回顾性地评价了CHA方法,证明与使用单一的3D药效团模型相比,使用多个蛋白-配体复合物的模型改善了虚拟筛选性能。相比之下,MYSPHASE通过聚焦在不同蛋白-配体复合物MD模拟过程中观察到的药效团特征来优化公共特征的药效团。在对PPARα 进行的回顾性评估中发现,该模型的性能优于使用X-射线衍射结构得到的3D药效团。
4.1.5 GRAIL (GRids of phArmacophore Interaction fieLds,2018)
药效团相互作用场网格( GRids of phArmacophore Interaction fieLds, GRAIL)法在MD模拟中以药效团水平描述MIF[77]。除了GRAIL外,该方法还为蛋白、水和配体(如果存在的话)生成了原子密度信息。GRAIL被应用于热休克蛋白90(heat shock protein 90)的MD模拟,结果表明药效团相互作用场可以帮助理解复合物配体的构效关系。
4.1.6 Water pharmacophore (2018)
与上述HSPR方法类似,水药效团(Water Pharmacophore,WP)方法旨在基于水合位点的热力学性质生成3D药效团[71,78]。通过联合使用热力学分析、MIF和分子对接等方法,WP在水合位点生成了水药效团。 但是,与HSPR相比,WP方法以高度自动化的方式生成单个3D药效团,所涉及的特征数量相对较少,从而在虚拟筛选中具有较高的性能。针对7个与药物相关的靶标优化了3D药效团生成的参数,作者能够为7个靶标中的4个产生了成功的3D药效团模型。
4.1.7 PyRod (2019)
与WP和HSRP相似,免费和开源软件PyRod专注于蛋白结合口袋中的水分子,以生成用于虚拟筛选的3D药效团[79]。然而,PyRod并不计算水合位点的热力学性质,而是基于快速计算的药效团启发的启发式打分函数来分析蛋白结合口袋中水分子的蛋白环境。这些信息被进一步处理,以动态dMIF的形式可视化结合口袋药效团特征,并生成用于虚拟筛选的药效团特征(图5)。由于仅在存在水分子的情况下进行打分,因此药效团特征优先放置于水占据率高的水合位点。这种水分子用配体片段代替后会导致熵增,因此增加了发现高亲和力配体的机会。与其它基于apo-蛋白的药效团方法相似,未经优化的3D药效团包含太多特征而无法进行有效的虚拟筛选。因此,用户必须首先基于dMIF及其在结合袋中的空间排列预先选择药效团特征。随后,将这种聚焦的3D药效团的特征组合生成基于用户自定义的特征(例如,独立特征或疏水相互作用特征等的最小与最大数量)生成药效团库。PyRod用一个Python脚本调用LigandScout对生成的药效团库进行评估[11]。根据回顾性评估中的ROC分析结果表明,PyRod的虚拟筛选性能在五个药物靶标中有三个优于对接方法,这为高效的虚拟筛选提供了一个直接可用的工作流。
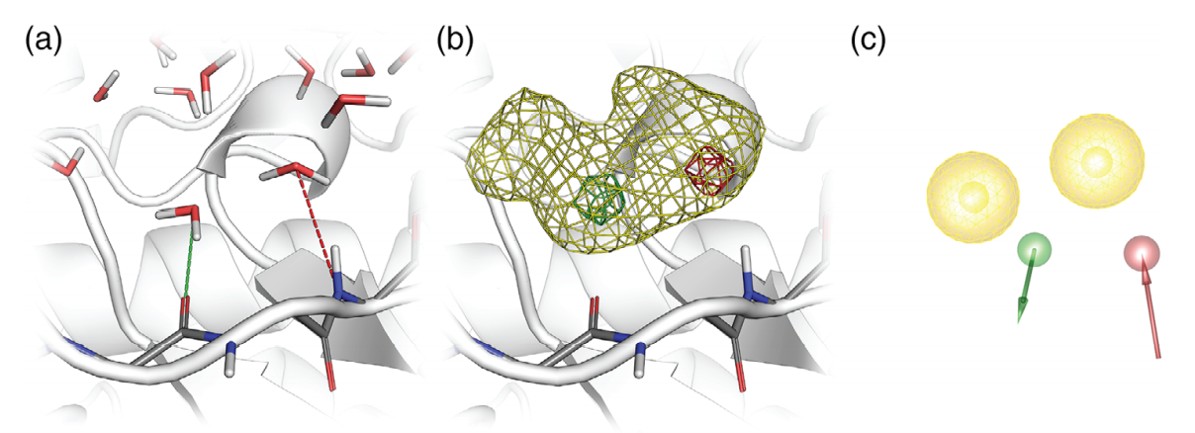
图5. CDK2结合口袋的PyRod分析(a) 分析水分子的蛋白环境,以生成(b)动态分子相互作用场(dMIFs)来描述结合口袋的特征,(c)dMIF又可转化为用来虚拟筛选的药效团特征:黄色为疏水相互作用,绿色为氢键供体,红色为氢键受体。
4.1.8 AutoDock Bias with Solvent Sites (2019)
AutoDock Bias with Solvent Sites(2019)使用基于混合溶剂的药效团将对接算法偏好于探针分子结合的热点,以提高虚拟筛选性能[80,102,103]。首先,作者对蛋白进进行了水和乙醇的混合溶剂MD模拟。接下来,对轨迹进行分析以识别乙醇羟基和甲基的结合热点。最后,将计算得到的乙醇热点的自由能作为AutoDock 4对接算法的一个能量项[28,80]。回顾性地评估了有偏的对接性能,结果表明,在大多数研究的测试体系上,与标准对接程序相比其性能得到了改善[103]。
4.1.9 Pharmmaker (2020)
Pharmmaker(2020)分析混合溶剂模拟以生成用于虚拟筛选的3D药效基团[81]。在本案例研究中,混合溶剂模拟使用了6种不同的探针分子,随后用DruGUI不同结合位点的成药性[104]。随后,对最具成药性的结合位点用Pharmmaker处理,选出具有高探针特异性亲和力的蛋白残基并识别出在蛋白残基与探针分子之间最频繁相互作用的快照。最后,用选定的快照生成3D药效团并将其用于Pharmit [89]的虚拟筛选,Pharmit是本文稍后会描述的一种用于药效团虚拟筛选的Web应用程序。
4.1.10 与系综对接(ensemble docking)的比较
除了3D药效团建模外,还经常讨论多个蛋白构象在提高分子对接性能方面的益处[105]。然而,采用多个X-衍射结构的系综对接结果表明,仅一些测试体系的筛选性能得到微小改善,这很难证明增加的计算成本是合理的[106]。蛋白构象聚类是降低计算成本的一个解决方案, 但未能从MD模拟中识别出最合适的蛋白构象[107]。与分子对接相比,当前的3D药效团方法比传统的静态药效团模型显示出明显的优势。例如,Dynopores以体积特征密度云的形式提供了相互作用的统计学特点。摆脱了传统药效团特征的球形特性,并将药效团特征表示为概率密度函数,这为提高虚拟筛选性能提供了绝好机会。此外,像PyRod这样的方法还考虑了配体结合的熵贡献,而熵用静态方法是很难评估的。与这些先进的药效团方法相比,系综对接(ensemble docking)是一种基于相同基础算法进行的计算昂贵的并行化。因此,MD模拟中包含的重要信息没有被系综对接适当地考虑,从而导致了相对较差的性能改进。
4.2 用药效团描述符训练机器学习模型
近年来,机器学习和人工智能在公共媒体中受到了极大的关注。算力的提高与可用数据的增加对现代药物发现过程产生了重大影响[108]。此外,3D药效团的概念也被用来开发几种新的机器学习方法。
4.2.1 HS-pharm (2008)
热点引导的基于受体的药效团(Hot-Spots-Guided Receptor-Based Pharmacophores,HS-Pharm)方法训练机器学习模型以减少基于apo的3D药效团模型的特征数量[30]。对~3500个可分辨的蛋白-配体复合物结合腔进行了分析,将超过600K的原子分为两组:相互作用组和非相互作用组。基于原子的空腔指纹是从收集到的空腔原子产生,数据的数据是关于药效团和扭转角特性、所涉及的残基及其蛋白环境的数据。这些指纹用决策树和贝叶斯分类器进行了训练与测试,以用来探测对配体结合重要的空腔原子。根据富集和ROC分析对方法的评估结果,可以确定性能最好的方法为随机森林决策树。最后,将这种方法应用于三个药物靶标,结果表明三分之二的3D药效团其性能优于对接方法。
4.2.2 Pharm-IF (2010)
将基于药效团相互作用指纹(pharmacophore-based interaction fingerprint,Pharm-IF)作为几种机器学习算法的输入,并评估其对小分子的对接结合模式(docking pose)进行排序的能力[82]。用五个药物靶标所有可用的原子模型生成了相互作用指纹,其编码了相互作用模式的类型和距离。这些数据随后用于训练几种机器学习算法来对已知的活性化合物和诱饵化合物(decoy)的对接结合模式进行排序。在一项回顾性评估中,Pharm-IF指纹与支持向量机结合显示出最佳的富集能力,其性能优于其他机器学习算法与对接打分函数。与使用PLIF(一种在MOE中实现的蛋白-配体相互作用指纹)相比,使用Pharm-IF训练的机器学习算法可以获得更好的富集性能[13]。与Pharm-IF相比,PLIF这种指纹不编码距离,这表明距离对成功预测的重要贡献。有趣的是,在对五个以上晶体结构的学习使模型能够比分子对接打分值更好地预测本研究所有靶标的活性。
4.2.3 DeepSite及其相关软件(2017)
DeepSite软件采用卷积神经网络(通常用于分析视觉图像的方法)以预测蛋白结合口袋的成药性[83]。约7,000种蛋白-配体复合物结构的蛋白用网格点覆盖,给每个格点指认了基于原子的药效团描述符。随后,将这些网格划分为子网格,如果它们的几何中心在结合位点几何中心的4Å之内,则将其标记为结合位点。由药效团描述符的3D网格表征的结合口袋的3D图像用于训练卷积神经网络。结果发现DeepSite结合腔检测的性能优于其它最新检测算法。后来,也使用类似的方法将药效学描述符用来训练卷积神经网络以预测小分子的结合亲和力(比如KDEEP[84])并用于指导新型分子的设计(比如LigVoxel[85]与LigDream[86])。所有上述方法都在本地安装的软件包中实现,但也可以免费用作Web应用程序。
4.3 应用3D药效团的网页应用
尽管Web应用程序不一定代表3D药效团概念本身的进步,但它们确实提高了可用性,从而将3D药效团带到了互联网时代。由于不需要在本地安装软件,并且所提供的Web应用程序都可免费用于学术研究,因此用户不仅规避了软件许可费并且无需高性能计算资源就可以对具有数百万个分子的数据库进行筛选。3D药效团搜索因而被更多用户所使用。
4.3.1 PharmaGist (2008)
PharmaGist Web应用程序允许用来生成基于配体的3D药效团[87]。程序对每一个提交的配体分析了可旋转键,这对于柔性分子的叠合很重要,这对用于与参比配体的叠合比对的药效团特征也很重要。程序会生成用户指定的最大数量的3D药效团,并且可以下载输出结果,其包含有3D药效团以及叠合好的分子。
4.3.2 PharmMapper (2010)
PharmMapper Web应用程序可用于小分子的靶钓(靶标预测),这对小分子的脱靶预测与多重药理学研究很重要[88]。采用基于蛋白-配体相互作用的方法或采用空腔检测算法识别的潜在别构结合位点的方法,从大约23,000个PDB晶体结构中生成了53,000多个3D药效团模型。提交的分子与所有保存的3D药效团模型进行匹配,匹配度作为小分子的打分值。
4.3.3 Pharmer-based web applications (2012)
Pharmer虚拟筛选软件[10]已用于多个Web应用程序,使得对几个小分子数据库进行高效虚拟筛选成为可能。AnchorQuery是专门用于识别蛋白-蛋白相互作用抑制剂[90]。用户上传蛋白-蛋白复合物,并指定可能对蛋白-与蛋白相互作用很重要的锚残基。该锚残基将作为3D药效团的一部分,可用于筛选数百万个可合成的小分子化合物库。ZINCPharmer可用于对ZINC数据库进行虚拟筛选,这是一个免费的虚拟库,收集了来自不同供应商的市售化合物[91,109]。该Web应用程序支持导入3D药效团,但也可用于从头开始生成的3D药效基团模型。最后,Pharmit可用于对多个商业供应商以及其它非商业数据库(包括ChEMBL和PubChem在内)进行虚拟筛选[89,110,111]。3D药效团可以由蛋白-配体复合物或配体设计得到。此外,还支持以几种不同格式3D药效团模型的导入。
5. 结论
在本文中,我们概述了3D药效团的原理及其在药物发现中的作用。3D药效团模型具有通用性、可编辑性和全面性的这一事实使它们可以应用在不同的场景。
一个主要的应用领域是通过虚拟筛选来识别新配体。 为此,3D药效团模型成为既可基于配体又可基于结构方式应用的唯一技术。无论哪种方式,3D药效团模型在计算上都非常高效,能够对非常大型的数据库进行虚拟筛选。抽象的化学官能团基本概念让骨架跳跃得以进行,并用于富集化学多样性的苗头化学物。总的来说,这使研究人员在可用数据、计算资源和测试能力方面有了更大的灵活性。我们选择的案例研究突出了基于药效团的虚拟药物筛选用于药物发现的能力,并显示了其对具有挑战性的靶标的适用性。而且,越来越受欢迎的基于片段的药物发现可以受益于药效团筛选,可大幅减少体外测试的片段数,理性地从片段母核进行片段生长[54,55]。
除了虚拟筛选之外,3D药效团还非常适合于研究、可视化类药分子的结合模式。它们由有限的化学定义的相互作用特征组成,这使得它们直观并易于理解。这在跨学科项目中是一种重大的优势,因为3D药效团模型能够合理地解释各种药理作用。为此,通常将3D药效团与其他方法(例如对接,MD模拟或机器学习)结合使用。从该领域精选的案例研究强调了3D药效团从机理上解释、理解蛋白功能的能力。此外,3D药效团还是研究人员之间进行交流的出色工具,而这一要素常常被低估。
然而,除了上述优点和可能性之外,经典的3D药效团模型也有某些缺点。 它们用静态模型表征高度动态的体系,并且它们的相互作用特征限于简单的几何形状(例如球形特征)。 此外,它们与其它模拟技术有一个共同的缺点:都专注于评估分子相互作用的焓,但在描述熵效应方面却不理想。然而,焓和熵都对配体与大分子结合的自由能的变化有贡献。尽管3D药效团生成的基本概念及其在虚拟筛选中的应用在过去30年中没有改变,但该领域仍有旨在解决这些缺点的各种进展。
因此,3D药效团模型与MD联合使用是一个具有巨大潜力的必然发展。 在本综述中,已经报道并描述了将MD集成到3D药效团建模中的不同方法[71–75,77–79,81,103]。然而,仅Dynophore方法是全自动的方法,它一次性解决了经典3D药效团的两个缺点 [74]。Dynophpre摆脱了传统球形几何结构的药效学特征可视化方式,用统计数据报告了MD轨迹中特征的出现频率以及不同的结合模式,揭开了配体结合的新前景。将这些性质-密度函数直接用于虚拟筛选代表了3D药效团建模中的真正范式转变。
几种先进的方法还考虑了3D药效团建模中配体结合的熵效应[71-73、78、79、81、103]。例如,PyRod分析了在MD模拟中水分子的蛋白环境,从而可以将药效团特征放置于具有特定热力学特征的水合位点上 [79]。这些水合位点可能在高度疏水的蛋白环境中藏匿水分子,或通过氢键以及结合口袋的形状严重约束水分子。将3D药效团限制在熵和焓的重要位点上的方法是很有价值的虚拟筛选工具,尤其是对于那些从apo结构生成3D药效团的方法。重要的是,PyRod是一种免费、开源的工具,可让该策略得到广泛的应用。
3D药效团概念与机器学习/人工智能的结合还处于起步阶段。尽管已经有一些方法[30,82-86],但我们预测将会有越来越多的研究和方法旨在使用药效团特征作为描述符或尝试从大数据中生成3D药效团。我们观察到提供免费的、基于药效团的虚拟筛选网络服务已形成一种新趋势[87-91]。
3D药效团领域的最新发展令人鼓舞,使用3D药效团的机会日益增长,其使用场景也日益具有挑战性,例如多靶标预测、结合动力学的模拟以及通路特异性受体激活。总的来说,3D药效团是计算机辅助药物设计工具箱的重要组成部分,非常适合于识别新型配体并理解其与大分子靶标的相互作用。


