
摘要:对于蛋白-配体相互作用模式的研究,目前有各种各样的采样方法允许快速访问大规模数据。主要的例子是大量使用分子动力学模拟应用于晶体结构,提供关于蛋白-配体复合物中结合相互作用的动态信息。从这些模拟提取的基于化学特征相互作用的药效团模型最近与一致性分方法一起用于识别潜在的活性分子。虽然这种方法快速并且可以完全自动化地进行虚拟筛选,但来自此类模拟的其它相关信息仍然不透明,并且迄今为止尚未完全开发出此类方法的全部潜力。为了解决这些问题,我们开发了药效团模型的分层图表示 (hierarchical graph representation of pharmacophore models, HGPM)。这种单一图形表示使得从长MD轨迹直观地观察大量药效团模型成为可能,并进一步强调它们的关系和特征分级。由此产生的交互式描述提供了一个易于理解的工具,可用于药效团选择,并为分析药效团特征组成和虚拟筛选结果提供视觉支持。此外,该表示可以适用于包括涉及相同蛋白和多个不同配体之间相互作用的信息。在这里,我们描述了从人葡糖激酶蛋白与变构激活剂复合物的两个X-射线共晶结构的MD模拟开始,HGPM的生成、可视化和使用。结果表明,大量的药效团及其关系可以以一种交互、高效的方式进行可视化,识别出独特的结合模式,并且从长MD模拟中得到的模型组合可以策略性地对虚拟筛选活动进行优先级排序。
原文:Arthur, G.; Oliver, W.; Klaus, B.; Thomas, S.; Gökhan, I.; Sharon, B.; Isabelle, T.; Pierre, D.; Thierry, L. Hierarchical Graph Representation of Pharmacophore Models. Front. Mol. Biosci. 2020, 7 (December), 1–17. https://doi.org/10.3389/fmolb.2020.599059. https://www.frontiersin.org/articles/10.3389/fmolb.2020.599059/full
编译:肖高铿/2021-01-03
前言
了解配体与大分子靶标的相互作用识别对于成功发现新型生物活性化合物至关重要(Fenwick et al.,2011)。 在药物设计中解决这个问题的一种方法是将配体-靶标相互作用建模为药效团。 人们将药效团定义为确保与特定生物靶标的最佳超分子相互作用和触发(或阻断)生物反应所必需的立体空间和电子特征的集合(Wermuth et al, 1998)。
一般来说,药效团模型要么源自配体-靶标复合物(基于结构)和/或一组已知的活性分子(基于配体),然后可用作计算机虚拟筛选 (VS) 的查询以找到具有相似立体电子特征的化合物(Langer,2010;Leach et al,2010;Schuster,2010)。基于结构 (SB) 建模的一个限制是靶标-配体复合物之间所有可能的相互作用可能无法捕获,因为它们来自静态表示。众所周知的一个事实是:蛋白是柔性的结构并且与配体的相互作用本质上是动态的,仍然是计算机解决方案中的一个重要问题(Cozzini et al,2008;Boehr et al,2009)。分子动力学 (MD) 模拟最近已用于对采集蛋白构象(Durrant and McCammon,2011 ;De Vivo et al,2016;Liu et al,2018 ),然后用于从最初的静态晶体结构推导出多个药效团模型。Choudhury等人(2015) 从 MD 模拟的每一个快照生成3D药效团模型,在对接与虚拟筛选重新打分后选择出性能最佳的模型。还可借助聚类(Sohn et al,2013;Spyrakis et al,2015 )方法来选择单个“表现最佳”的药效团模型,提供比“经典”的基于X-射线衍射晶体学结构更好的虚拟筛选结果药效团模型。但是,要确定“性能最佳”的模型,需要已知活性和非活性化合物的数据集来评估模型的性能。在新靶标早期苗头化合物发现阶段,此类信息可能尚还缺乏,用虚拟筛选来对药效团模型进行优先级排序具有挑战性。
为了克服选择一组独特的代表性药效团模型的需要,Wieder 等人 (2017) 开发了“Common Hits Approach” (CHA),其中从 MD 模拟得到的多个 3D 药效团模型根据它们的特征组成进行分区,并用于后续的 虚拟筛选。使用一致打分函数来对初始的每个代表性模型获得的筛选结果进行排序和组合,从而能够基于一组MD衍生的模型对虚拟命中化合物进行优先性排序,从而获得单个最终的命中化合物列表。最近,Polishchuk等人(2019)改进了工作流,通过调整一致性打分函数使得虚拟筛选搜索到的每个分子的构象数量也得以考虑。基于这些研究,Madzhidov等人(2020)分析了一组药效团模型的性能,并为一致性打分开发了一种的概率方法,从而得出了一种对池中表现表现不佳的模型不太敏感的方法。尽管这些基于一致性的方法比“经典”的药效团方法提供了更好的结果,但由于需要多次运行虚拟筛选,此类方法需要大量的计算资源。
如今,MD 模拟可以对构象空间进行彻底地采样——即使是大型生物体系,基于结构的药效团模型的生成不再局限于单晶结构。作为现代硬件改进的直接结果,大多数计算实验室现在可以在几小时内执行纳秒级的MD模拟。然而,对非常大的库(数百万化合物)执行连续虚拟筛选仍然是一个关键的时间限制因素。为了解决这个问题,本文提出了药效团模型的分层图形表示( hierarchical graph representation of pharmacophore models,HGPM),其目的是便于理解药效团模型相关信息的可视化,从而有助于为后续处理步骤确定药效团模型的优先级排序和选择。虽然以往的工作通过对晶体结构或3D药效团信息进行聚类以减少了药效团模型的数量,但是此形图表示侧重于分层药效团特征信息的视图,以支持用户在模型选择过程中减少随后跑虚拟筛选的模型数量。例如,源自 MD 模拟的多个药效团模型的单一表示具有以下几个优点:(i) 引入易于理解的基于图形的视图,用于观察观察到的所有独特模型及其关系(Maggiora and Bajorath, 2014; Métivier et al, 2018)。 (ii) 更简单、更不容易出错的 3D 药效团模型选择过程,特别是对于虚拟筛选运行的长时间MD模拟。 (iii) 通过添加从其他体系或MD模拟生成的模型来扩展显示信息的可能性。
以下各节将专注于算法细节和用于生成药效团模型的分层图形表示的计算过程。此外,还以人己糖激酶IV作为案例研究的进行演示和讨论,说明如何用该方法展示并充分利用从MD模拟中获得的药效团信息。
材料与方法
蛋白-配体复合物结构准备
从RCSB PDB数据库(Berman et al,2000)下载了与激活剂复合的人葡萄糖激酶的两种晶体结构,PDB ID 为 1v4s(Kamata et al,2004)和 4no7(Petit et al,2011)。使用 RCSB PDB 比较工具(Prlić et al,2010 )和 jFATCAT_flexible 算法(Ye and Godzik,2003 ),对蛋白质序列进行比对并随后对氨基酸重新编号。4no7 复合物中不存在氨基酸 92-99。由于它们在模拟过程中没有影响蛋白质稳定性,并且在1v4s体系中没有观察到与配体的相互作用,因此没有对它们进行建模。包含比对区块的表格可在补充图1中找到。Maestro软件 (Schrodinger, 2010) 用于去除水分子 ,加氢并对结构进行能量最小化。末端的封端、溶剂化和蛋白质复合物的离子添加通过CHARM-GUI 网络界面进行设置(Jo et al,2008)。有关蛋白-配体复合物准备的信息可在补充表1中获得。
分子动力学模拟
MD模拟是使用Amber 16进行的(Case et al,2016)。配体的参数是用tleap生成使用通用AMBER力场 (GAFF,Wang et al., 2004)。MD 模拟流程从125 ps的平衡和加热阶段开始,时间步长为1fs。然后在 303.15 K的温度下使用Langevin动力学对每个体系进行总共300ns的模拟,由3个100 ns的重复组成,各自不同的初始速度。使用Monte Carlo 恒压器将压力保持在 1 个大气压左右。SHAKE 算法(Ryckaert et,1977 )用于保持所有涉及氢原子的键保持刚性。生产运行的时间步长设置为 2 fs。蛋白质及其配体的均方根偏差图见在补充图2。
库的生成
在人类葡萄糖激酶上测量的实验活性的化合物取自 ChEMBL 数据库(Gaulton et al,2017)。总共提取了756个对靶蛋白具有活性(表达为EC50)的独特分子。该集合根据 1.5 μM 的活性阈值进行拆分,601 个分子标记为活性分子(actives),155 个分子标记为诱饵(decoys)。 KNIME 分析平台(Berthold et al,2009)与 InteLigand Expert KNIME LigandScout Diversity Picker 节点(InteLigand Expert KNIME Extensions)结合使用,基于ECFP相似性从ChEMBL库中提取了20,000个多样的分子,也标记为诱饵。最后,使用LigandScout 4.4 Expert(LigandScout 4.4 Expert)中的idbgen算法计算了一个虚拟筛选库。该过程包括使用icon Fast设置(Poli et al,2018 )为 20,756 个分子中的每一个生成最多 25 个构象。活性分子根据 ECFP 相似性聚类分为 5 组。每个簇中的代表性分子的示例见补充表 2。来自X 射线衍射结构PDB 1v4s 和 4no7 的配体分别在簇号 4 和 2类中。
药效团生成与虚拟筛选
使用LigandScout 4.4 Expert(Wolber and Langer,2005)为 MD 模拟的每一帧输出生成基于结构的药效团模型。LigandScout生成的模型支持以下化学特征类型:疏水相互作用、氢键供体/受体、正/负离子化区、芳环和卤键供体特征。此外,还创建了来自 1v4s 和 4no7 的 X 射线衍射晶体结构的药效团模型。在模型生成之前删去水分子。 LigandScout的Activity profilingKNIME节点用于将20,756个分子的专用数据库用各个药效团模型进行虚拟筛选。每次虚拟筛选都用生成ROC曲线并计算在数据库分子总数特定百分比时的ROC曲线下面积 (AUC)来评估模型的性能。
药效团模型分层图的生成
特征向量(Feature Vectors)与图节点(Graph Nodes)
从MD模拟得到的药效团模型以Wieder等人(2017)论文所述的相关方式转换为特征向量。向量的每个元素代表了在体系中观察到的一个独特的药效团特征。向量的每个元素代表了一个在体系中观察到的独特药效团特征。在本研究中,如果药效团特征在以下任何成分(药效团特征类型、配体标识符和/或相互作用环境残基的标识符)中存在差异,则其被认为是独特的。在识别独特的药效团特征时不考虑3D信息。因此,例如,对于与丝氨酸相互作用的配体氮原子产生的氢键受体特征将被认为不同于代表与苏氨酸相互作用的相同氮原子的相应特征,而不管它们的共享特征类型或它们的3D位置。两种药效团特征都将被认为是独一无二的。也就是说,独特的特征是特定于它们所创建的药效团集的,特征矢量也是如此。特征向量表示为描述3D药效团模型组成的位串(bit-sting):0值仅表示模型中不存在所考虑的独特特征,1 值表示该特征存在。位串表示允许快速过滤具有相似特征集的药效团模型,此外还可以在模拟过程中快速计算特征出现计数。分层图(hierarchical graph)表示中的每个节点都与一个独特的特征向量相关联,并包含额外的派生信息,例如相关的帧数、出现次数和链接的药效团模型。图 1 显示了一组药效团模型的特征向量生成过程及其与图节点的关联。为了限制初始药效团模型中的噪声,根据它们的出现次数过滤独特的特征向量。对药效团模型进行过滤,以保持模型的出现至少是Wieder等人(2017)论文所述的2次,或初始帧数的0.001倍。在MD模拟过程中观察到的第一个药效团模型,针对分层图中的每个唯一特征向量,都可考虑用于虚拟筛选。
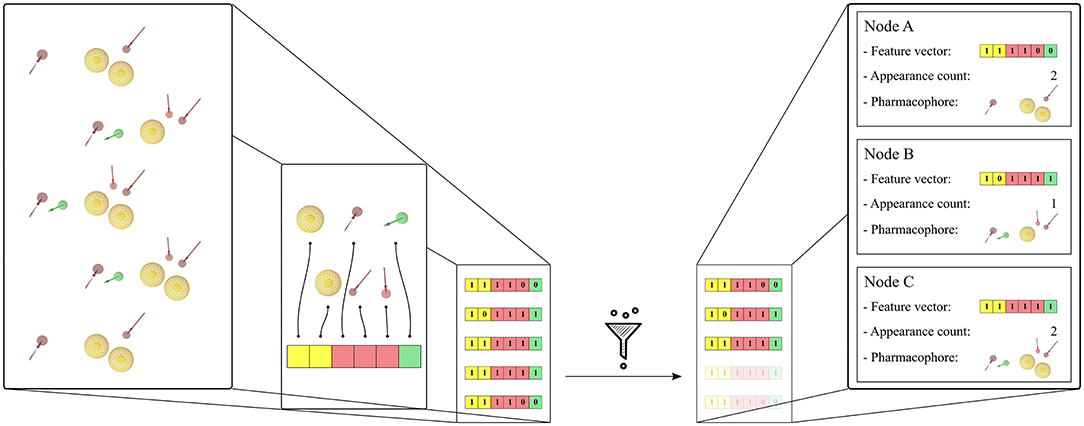
图 1. 从5个药效团模型的初始集合中生成特征向量及其节点表示。首先识别所有遇到的独特特征,将一组药效团模型转换成一组相应的特征向量集。然后,根据药效团模型中相应独特特征的存在与否,特征向量元素被初始化为1或0。执行过滤步骤以删除具有相同特征向量的重复项。最后,为每个独特的特征向量创建图节点,并存储相应的药效团模型和出现计数值。药效团模型特征:黄色球体(疏水性)、红色和绿色箭头(分别为氢键受体和供体)。 相应的向量特征被相应地着色为黄色、红色和绿色。
分级链接(Hierarchical Linkage)
图的分级链接基于每个节点中特征向量的独特药效团特征组成。如果节点的特征向量是彼此的子集或超集,则在节点之间创建链接。图 2 描述了链接过程。如果两个特征向量不表现出子集或超集关系,则临时创建一个新的特征向量。这个新的特征向量表征两个考虑节点的独特特征的交集。如果这个临时特征向量与一个已经存在的节点等同,那么两个考虑的节点将链接到这个节点。如果临时节点是唯一的,则会创建一个新的永久节点。已经实现与新节点相关联的“非自然存在”的特征向量的创建,以允许生成唯一的分层图。因此,特征向量的“观察到的”或“非自然存在的”性质以其出现次数的形式存储为每个图节点的属性。一旦生成了所有子集和超集链接,就删除冗余路径。
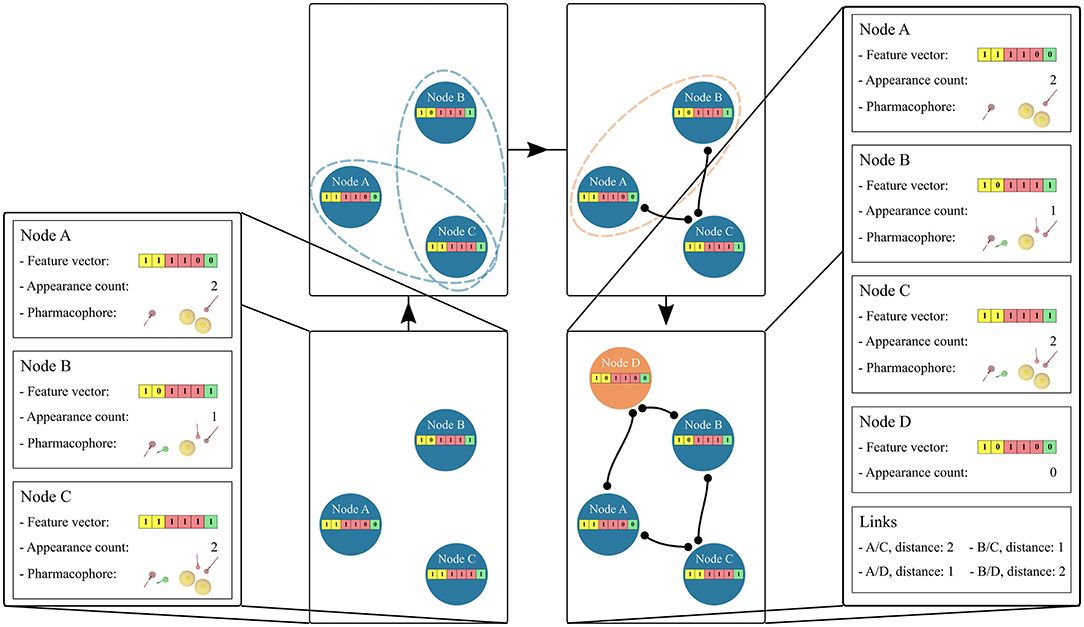
图2. 图节点的分级链接。从蓝色的3个“观察到的”节点开始,测试存储的特征向量的子集/超集关系,用蓝色虚线椭圆表示。如果找到该关系,则创建边。在两个节点没有描述这种关系的情况下,用橙色椭圆表示,创建一个新节点并将其链接到它们。将此“非自然存在的”节点的出现计数设置为0,并将其颜色更改为橙色。药效团模型特征为:黄色球体(疏水性)、红色和绿色箭头(分别为氢键受体和供体)。相应的向量特征分别着色为黄色、红色和绿色。
可视化
为了轻松理解图形节点及其链接中包含的信息,可视化起着至关重要的作用。从特征向量开始,关于药效团模型的组成以及它们之间的分级链接的信息就已经存在。几个可视化参数可用于描绘额外的图形属性。本文已选择以下属性,除非另有说明:
- 药效团模型的出现次数用节点的大小表示。因此,出现次数越高,节点的视觉表现就越大。
- “观察到的(Observed)”或“非自然存在的(Artificial)”节点的性质用其颜色表示。蓝色表示“观察到的”节点,橙色表示“非自然存在的”节点。
- 根据组成药效团的独特药效团特征的数量,在 x 轴上组织节点以表示该药效团模型的特异性。考虑一个节点,同一列中的每个其他节点具有相同数量的药效团特征,其左侧的每个节点由较少的特征组成,而其右侧的每个节点由更多的特征组成。
- 用多维缩放方法 (Multidimensional Scaling Method,MDS) (Mead, 1992; Borg and Groenen, 2003)进行降维来表征药效团模型的相似性。 这种方法将距离矩阵的所有元素放在一个维度中,尽可能地保留节点之间的距离。 通过计算节点特征向量之间的曼哈顿距离来获得距离矩阵。然后,药效团模型之间的相似性由投影在图的垂直轴上的节点之间的相对距离表示。 通过显示缩放数据的方差比例来可视化该过程的可靠性。
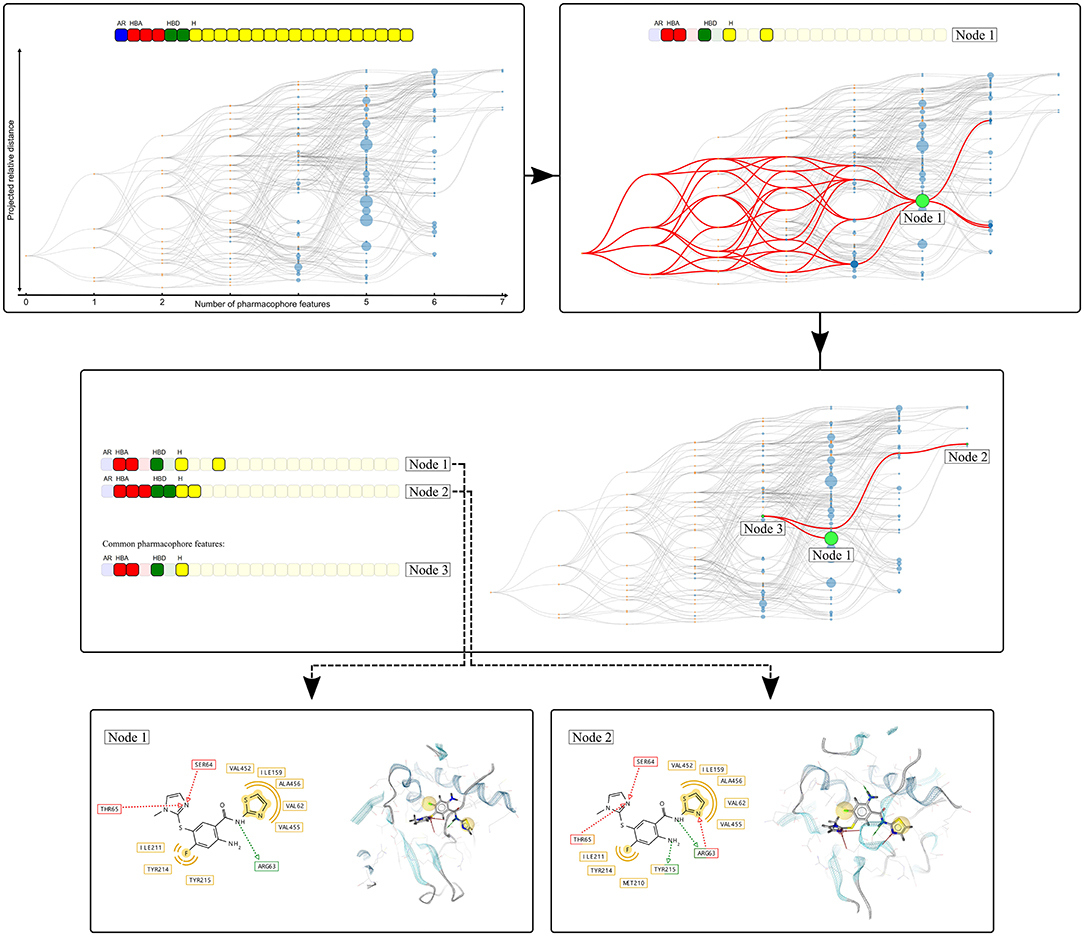
图3. 从人葡萄糖激酶与激或剂(PDB 1v4s)复合物的分子动力学模拟得出的药效团模型的分层图表示的可视化。特征向量呈现在图的顶部,每个框代表一个独特的药效团特征。框的颜色表示相应特征的类型:黄色(疏水性)、红色和绿色(分别为氢键受体和供体)和蓝色(芳香性)。特征向量下方的分层图表示在模拟过程中观察到的所有药效团模型。节点通过分级关系链接,颜色表示起源:蓝色(“观察到的”药效团模型)或橙色(“非自然存在的”模型仅由“观察到的”药效团的特征子集组成)。该图是交互式的,选中一个节点可绘制出所有相关的药效团模型,如选中节点1所示。当选中两个节点时,描绘药效团特征交集的节点(节点3)会被高亮显示,正如选中节点1和2所描绘的。对于每个节点,可以轻松检索相关的药效团模型。
结果与讨论
案例:葡萄糖激酶
己糖激酶IV或葡萄糖激酶 (GK) 是负责葡萄糖磷酸化的同工酶 (Beck and Miller, 2013)。血浆中葡萄糖的浓度决定了GK在其活性和非活性状态之间的构象转换。葡萄糖水平对GK活性的影响使这种酶充当负责人体内葡萄糖稳态的传感器(Bell and Polonsky,2001)。因此,GK一直是开发抗糖尿病药物的主要目标(Kamata et al., 2004; Osbak et al., 2009; Petit et al., 2011)。
本研究选择了与激活剂结合的GK活性构象的两种晶体结构(PDB 1v4s 和 4no7)。 图 4 描绘了配体结构、在结合口袋中的位置以及来自独立X射线实验的药效团特征。这两个配体的结合模式相对于能够与精氨酸63的主链形成相互作用的药物受体氢键供体特征和受体特征显示出相似性。正如在之前的研究中观察到的那样(Petit et al,2011),残基 92-102组成的Loop是无序的,这导致变构子口袋的打开,在4no7 PDB结构中可以容纳配体的氯苯基甲砜。
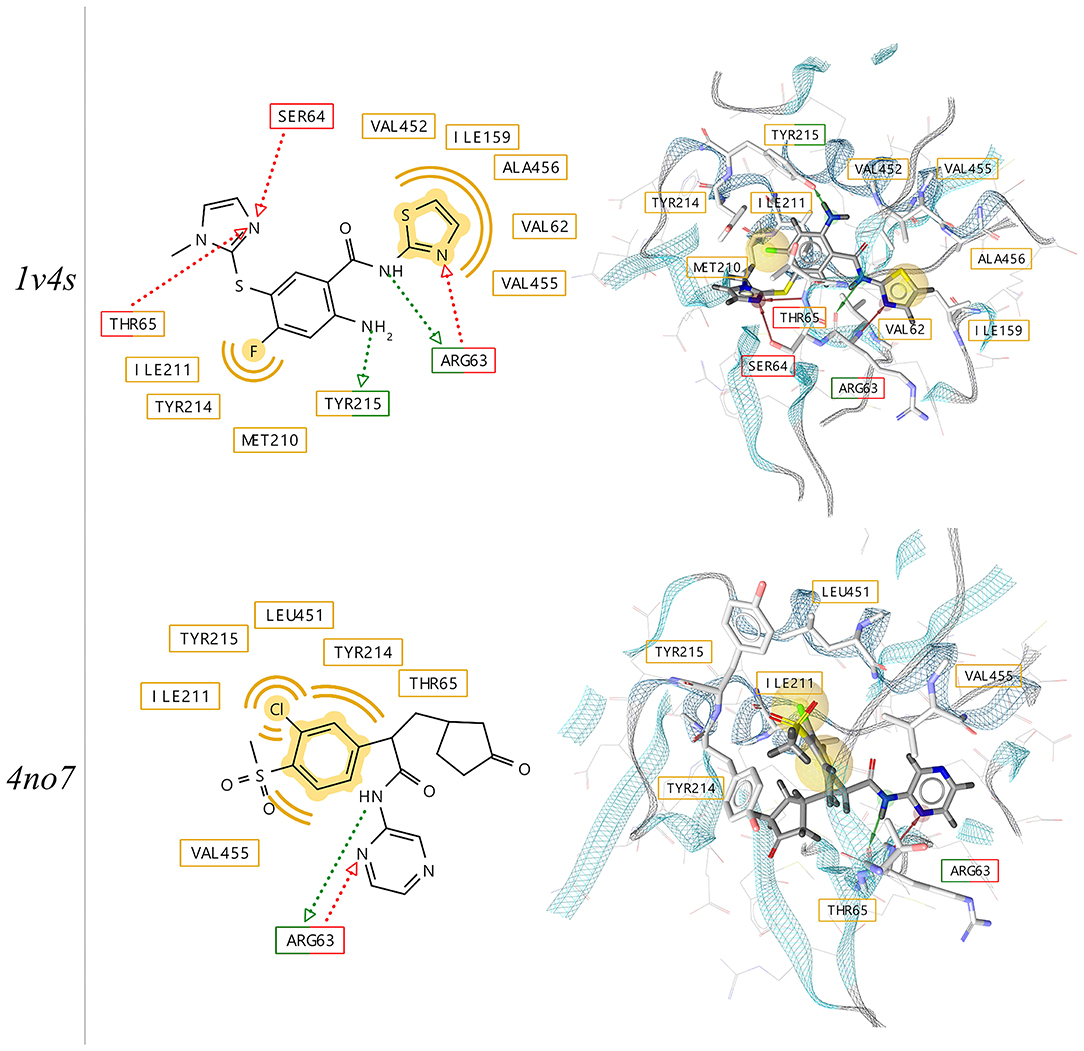
图4.使用 LigandScout 4.4分析PDB 1v4s 和 4no7得到共晶配体与人葡萄糖激酶的2D 和 3D相互作用示意图。疏水性、氢键供体和受体相互作用分别以黄色(球体)、红色和绿色(箭头)显示。
药效团模型的分层图(Hierarchical Graphs of Pharmacophore Models)
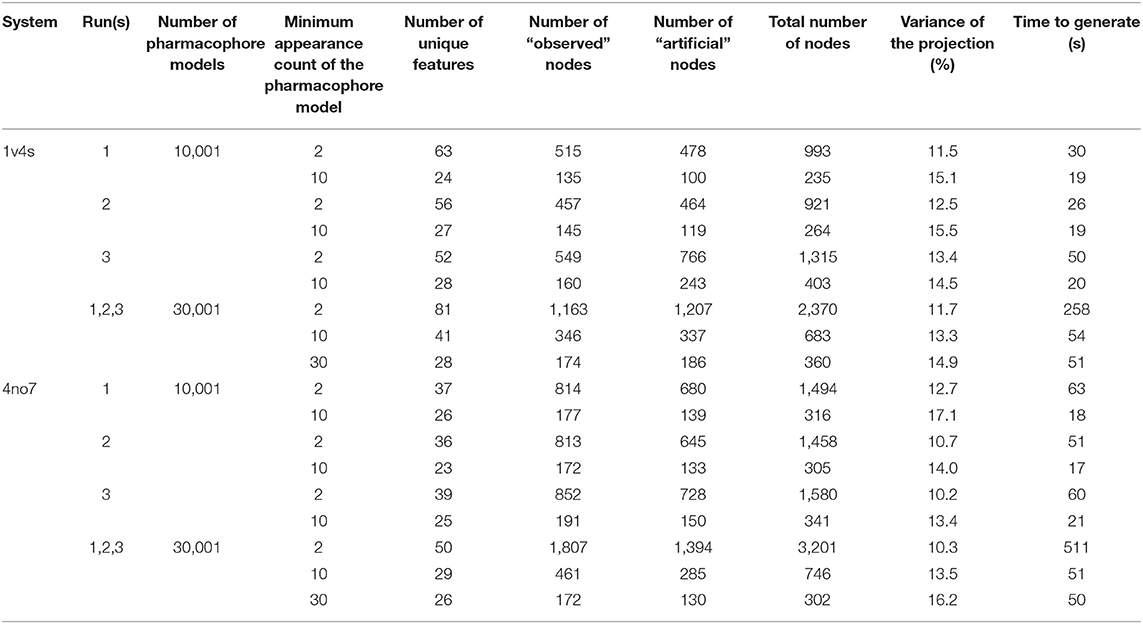
对两种蛋白质-配体复合物进行了三次MD模拟运行,每次100ns。从获得的MD模拟轨迹中,提取10,000帧并随后用于如方法部分所述的药效团模型的生成。 然后从基于框架的药效团模型(还包括基于晶体学结构的药效团模型)生成分层图。在对药效团模型进行分级层生成过程之前,根据它们的出现次数过滤药效团模型。为每个单独的计算以及为每个晶体结构的所有计算的统一生成图。表1总结了图的组成。在之前的文献(Wieder et al,2017)中,我们对独特的药效团模型出现次数使用了过滤标准,以便通过删除在模拟过程中只出现一次的药效团模型来降低噪声。在本研究中,我们研究了几个值对过滤标准的影响,丢弃出现在小于2帧到小于1%帧的模型,以便既减少噪声又提高图的可读性。用于图的生成和可视化的源代码可在线获得(HGPM实施的源代码,2020)。由于知识产权原因,LigandScout药效团模型的生成和处理源代码不在获得之列。还可以在线访问用于从4no7的第一次MD运行生成的药效团模型的分层图表示的交互式演示的输出数据(HGPM演示,2020)。当将鼠标悬停在特征向量上时,可以在在线演示中看到该体系所有独特特征的列表。
表1. 每个MD模拟的HGPM结果统计:图节点组成、药效团过滤标准、MDS投影的方差和生成时间。
For the investigation of protein-ligand interaction patterns, the current accessibility of a wide variety of sampling methods allows quick access to large-scale data.
独特特征向量的分析(Analysis of the Unique Feature Vectors)
为了可视化体系每次MD模拟所获得的药效团模型的组成,独特的观察特征的单独划分如图5的Venn图所示。对于1v4s体系,如果将出现次数过滤标准设置为2帧或更多,则在3个MD运行的每一个中存在81个(46%)唯一观察到的特征中的37个。当将出现次数标准设置为10帧或更多时,该比率保持相似,39个(46%)独特观察特征中的18个是共同的。 尽管总可观察到大约一半的独特特征,但是每个单独计算总会观察到仅存在于特定计算中的5-17个独特特征(2帧或更多的过滤标准)。当过滤标准设置为10时,仅在特定计算中观察到的独特特征的数量减少,但比率保持相似。
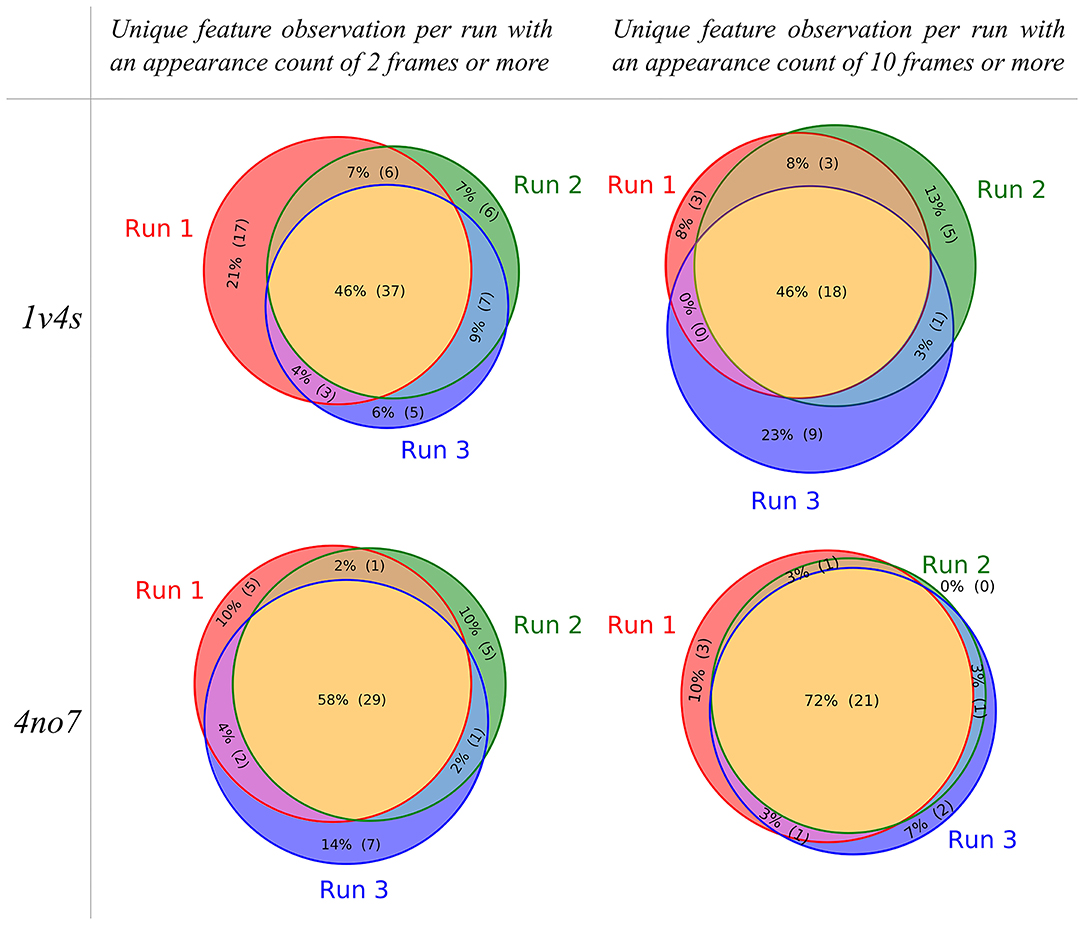
图5. Venn图显示了1v4s与4no7体系MD模拟之间的独特药效团特征相似性。根据出现次数标准对独特特征采样的药效基团模型进行过滤,左侧图设置为2,右侧图设置为10。
就4no7体系而言,趋势稍有不同,3次运行的共同独特特征比率的增加。当出现次数过滤标准设置为2帧或更多时,观察到在50个(58%)独特的共同特征中有29个;当过滤标准设置为10或更多时,在29 个中观察到 21 个 (72%)。因此,在过滤标准设置为 2 和 0-3 且标准设置为 10 的情况下,仅在单次运行中观察到的独特特征的数量减少为 5 到 7 之间。虽然无论运行如何,总是会存在几个独特的特征,每个单独的MD模拟均会提供独有信息。考虑从为每个体系执行的 3 次运行中的任何一次获得的每个药效团模型,进行了这项工作中提出的每一个进一步分析。本研究的每一个进一步分析,都考虑了每个体系进行3次运算,并从中获得的任意一个药效团模型。
应用于出现计数的过滤标准已被设置为降低层次图在节点和链接数量方面的复杂性,如表 1 所示。 为了研究过滤标准对特征向量组成的影响,图 6 描绘了独特药效团特征的性质细节。在 1v4s 和 4no7 的 MD 模拟中观察到的所有独特药效团特征中,81 个中的 62 个和 50 个中的 31 个是疏水相互作用。大量疏水特征部分是由于特征序列生成算法的性质造成的。例如,涉及1v4s中配体氟原子和氨基酸ILE211、TYR214、TYR215 之间的疏水特征被认为不同于涉及配体上相同氟原子的另一个疏水特征,但不同的氨基酸如THR65、MET210、ILE211、TYR214、TYR215,即使第一个特征的每个氨基酸也参与第二个特征。这是LigandScout对疏水特征的定义中固有的,并一直保持原样。
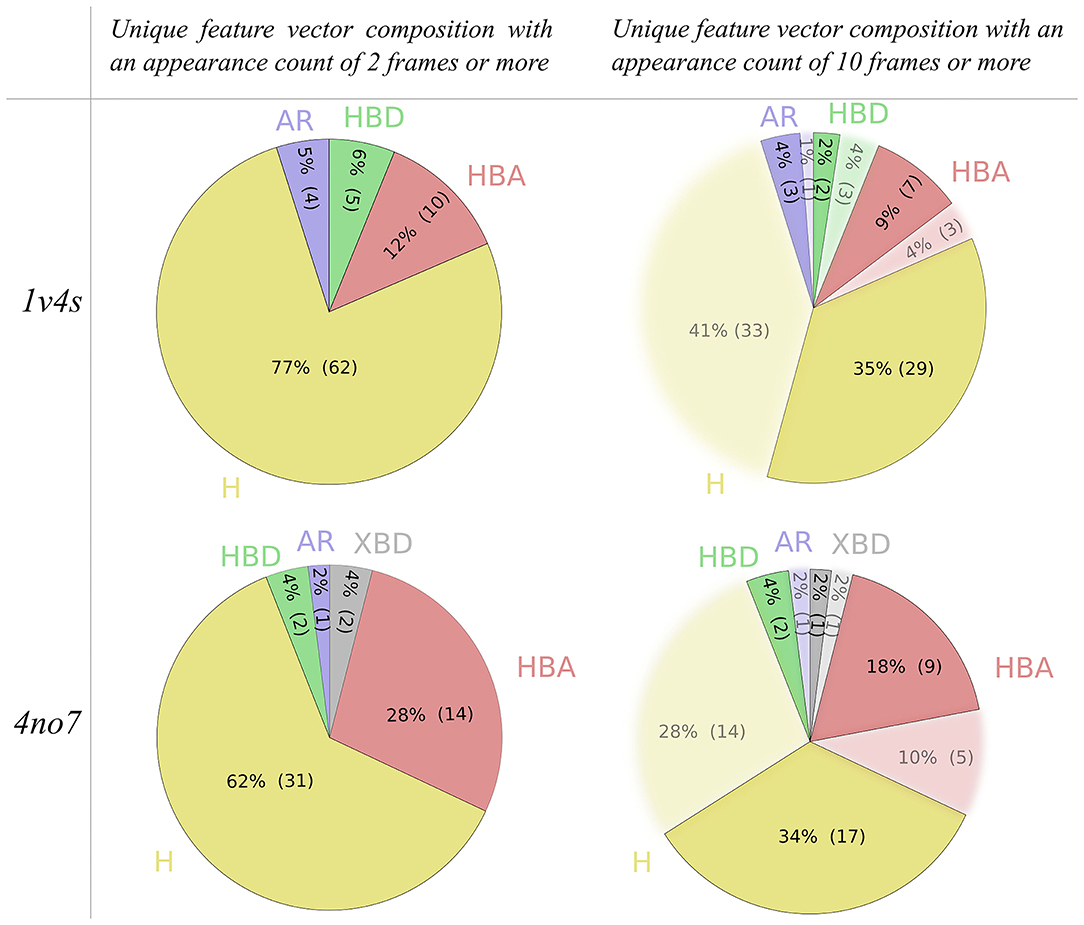
图6. 基于1v4s和4no7体系采用的出现次数过滤标准,3次MD模拟生成特征向量的独特药效团特征组成的饼图。 疏水相互作用为黄色,芳香相互作用为蓝色,氢键供体为绿色,氢键受体为红色,卤素键供体为灰色。
然而,将出现次数过滤标准设置为10帧或更多帧往往会丢弃比任何其他类型的药效团相互作用更多的疏水特征,如图6所示的两个体系中都可以观察到。表1还提供了出现次数过滤标准设置为帧数的1%的分层图信息。出于图可读性的原因,我们选择在本研究的其余部分使用为10的过滤标准。
分层图(Hierarchical Graphs)的使用与分析
图7展示了两个被研究体系的分层图。可视化部分详细描述了可以从这些图中直观地检索到的信息。
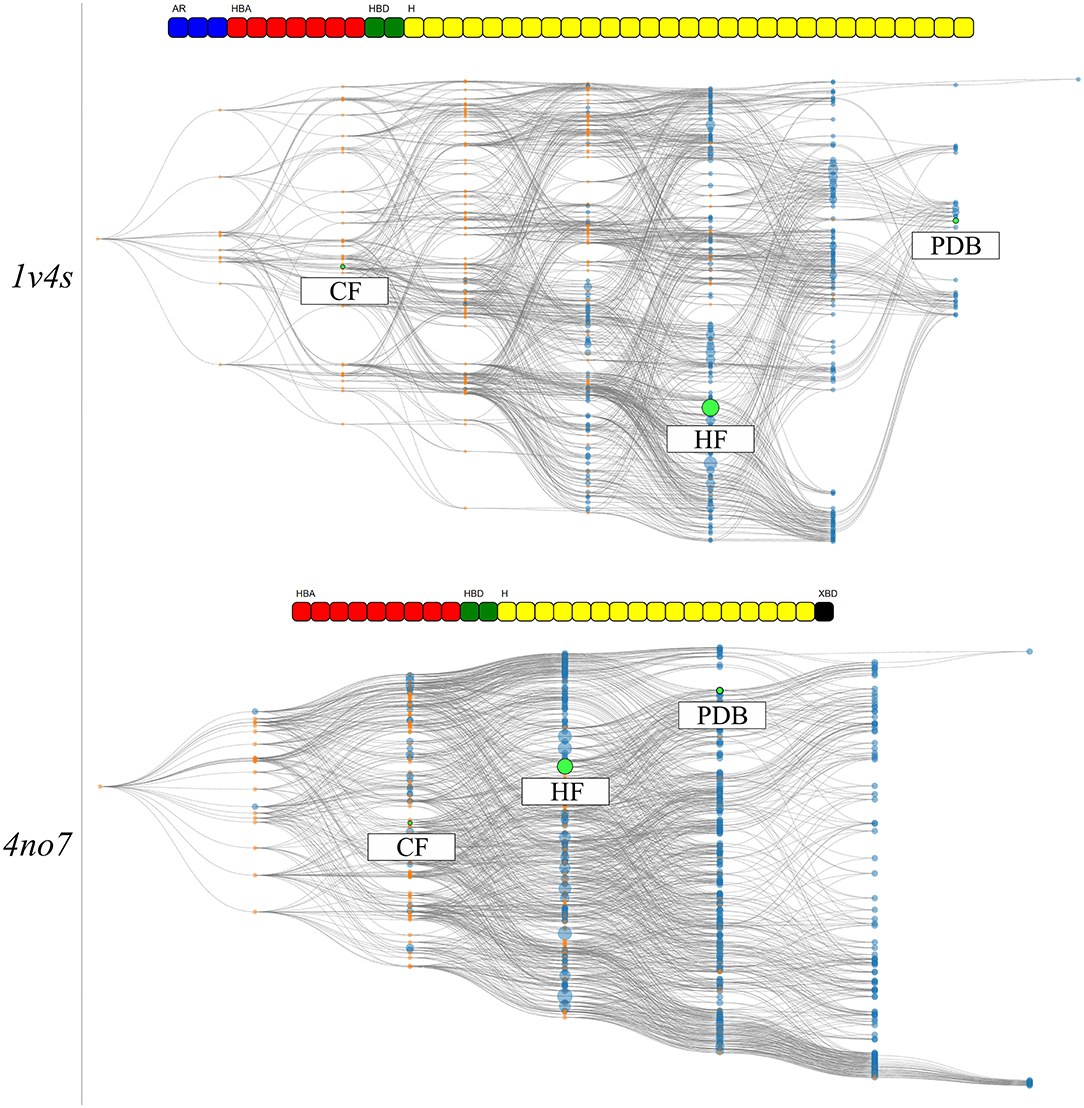
图 7. 3次 MD模拟运行的HGPM。顶部:1v4s体系;底部:4no7 体系。与从PDB结构获得的药效团模型相匹配的节点、出现频率最高的模型以及在体系晶体结构之间共有特征数最多的模型以绿色高亮显示,并将HF和CF分别标记出来。高亮显示的模型的特征向量显示在相应分层图的上方。
对于体系1v4s,该图有9列,因此包括具有多达8个药效团特征的模型。有几个节点的模型接近从晶体结构获得的药效团模型,标记为 PDB。然而,没有观察到其他药效团模型包含由 PDB 节点表示的特征超集。在3个运行中具有最高出现次数的节点(HF节点)从PDB节点垂直移位,虽然它们共享3个特征,但是在特征分级方面并不直接相关,如补充图3中更详细所示。与精氨酸 63 相互作用的氢键供体和受体特征相关的节点(CF节点)位于图的第三列,因其具有的特征数量较少,因此不专门说明。PDB和HF节点用于对活性和诱饵数据库进行虚拟筛选,如方法部分所述。此外,代表HF和CF节点超集的所有药效团模型用于跑一致性筛选。一种考虑到所有观察到的药效团模型的一致性方法CHA(Wieder et al,2017 )用来跑需要筛选。 虚拟筛选结果的总结见表 2。
表2.两个体系用不同药效团模型进行虚拟筛选的结果

我们观察到1v4s的PDB药效团在虚拟筛选时没有命中任何化合物,这可能是由于定义其特异性结合模式的氢键数量以及特定方向的氢键向量造成,7个药效团特征中有5个包含定义了方向的氢键。该体系的其它4个药效团性能表现良好,在1%数据库分子数量处的曲线下面积(AUC)大于0.96。 就1%的数据库分子水平AUC而言,HF药效团模型与其它模型一样性能表现良好,但略低于其它模型,并从20,756个分子中回收65个。就AUC而言,CF节点的所有超集模型具有最稳定和最好的结果,甚至优于CHA。良好的结果与用于筛选的大量药效团模型相关,其中236模型用于CF+的选择,346个模型用于CHA。最后,作为HF节点超集的所有药效团模型的集合性能与单独的HF节点近似,但搜索到87个分子。
4no7体系的药效团模型的分层图与1v4s体系的不同之处主要在于图右侧的两个分支涉及具有最高数量特征的节点。 此外,PDB、HF和CF节点在垂直方向上非常接近,并且在分级上相互关联,如补充图4所示。 这得出的结论是,这三个节点仅是最具特异性的药效团模型中的两个分支中的一个分支的一部分。 因此,选择了一个附加节点将第二个分支的任意模型包含进来,在图7中将其标记为“Selection”。表2显示了用PDB和HF节点获得的虚拟筛选结果。应用CHA,并考虑PDB、HF、CF以及自定义选择节点的药效团超集进行额外的一致性打分。对4no7,PDB药效团在数据库的1%处获得最好的AUC结果,并搜索到401个分子。 至于1%的AUC,CHA表现得与PDB节点一样好,并且与其他方法相比在高百分比时具有相当的AUC值,同时取回13,868个分子来,显示了其稳定性。HF节点模型的表现不如其它模型,在1%的AUC值为0.81,在5%时AUC值跌为0.41。然而,HF节点的所有超集模型比单独的HF节点获得显著更好的VS结果。最后,基于“任意”节点的选择具有高AUC值,在所有阈值都优于1%时观察到的最佳结果。有趣的是,“任意”选择的节点和HF节点具有不同的特征组成,但是仍然具有高AUC值。为了仔细检查这一观察结果,我们观察了通过这两种方法搜索到的活性分子的结构聚类,如补充图5所示。我们观察了截至2,076个分子(数据库的10%)的命中列表,任意选择的节点主要从簇号2、3和5搜索分子,而HF节点从所有的5个簇搜索相应数目的分子。虚拟筛选命中列表组成上的这种差异表明存在两种不同的结合模式,涉及活性分子的结构差异。因此,研究为什么这两个不同的分支仍然能够区分活性分子和非活性分子,对于阐明高亲和力GK配体的结合模式具有很高的价值。还可以强调的是,在对研究体系没有先验知识的情况下,与单个药效团模型相比,多个药效团模型被证明在AUC值方面特别稳定和可靠。
用虚拟筛选结果着色的药效团模型分层图的分析
分层图表示为药效团模型的选择和评估提供了直观的支持,如前一部分所述。尽管如此,可以描述附加信息以强调药效团模型的特殊特征。为了更好地理解正确区分活性分子和非活性分子所涉及的特征,所有与分层图的特征向量相关的药效团模型都用于虚拟筛选。然后,每个节点根据其在数据库的10%处的AUC值进行着色。对于这两个体系,相应的结果如图8所示。在所示的分层图中,节点越绿,其AUC值越接近1,其颜色越红,其AUC值越接近0。 “非自然存在的”节点不用于虚拟筛选,并显示为灰色。
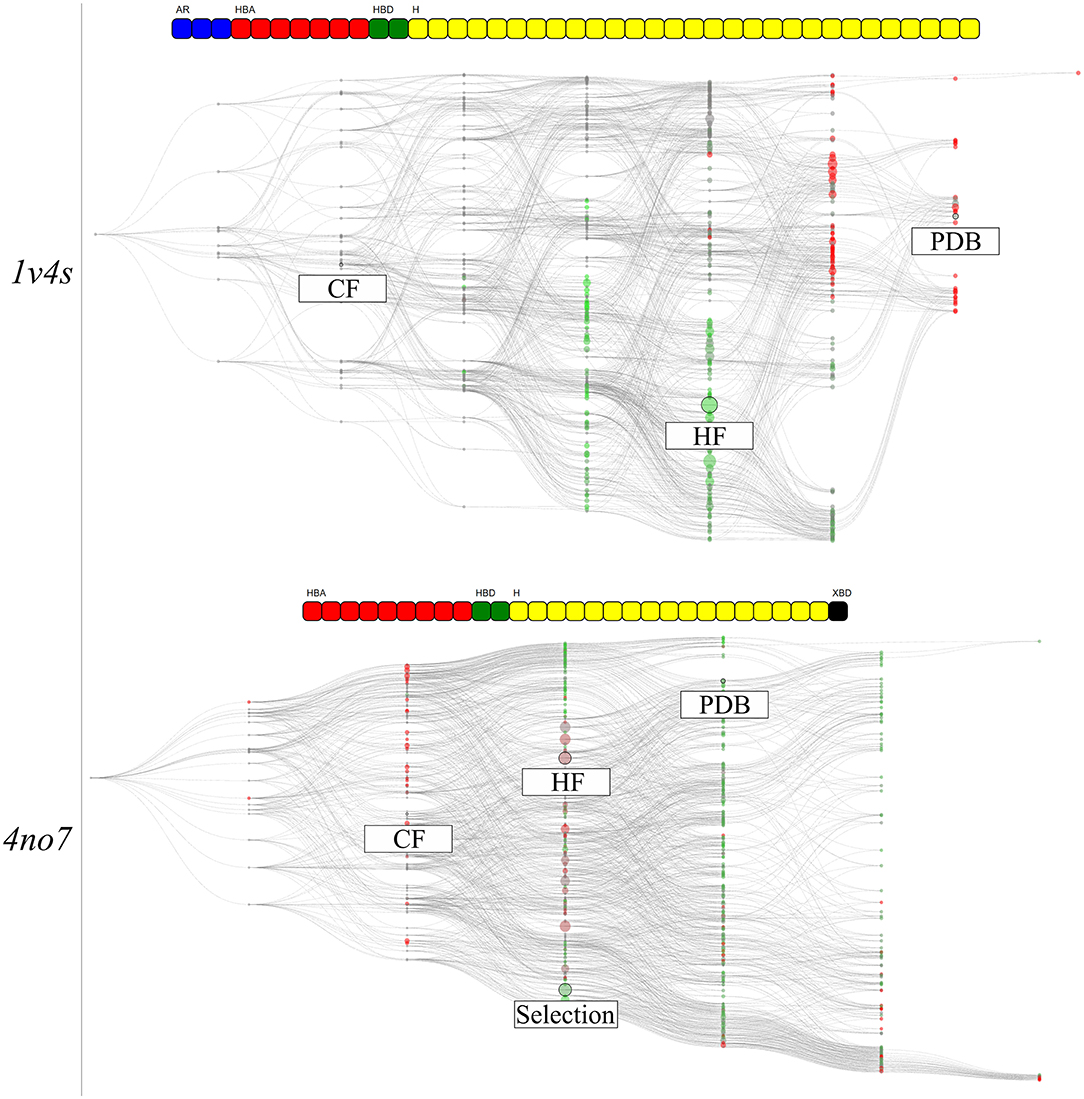
图 8. 上:来自PDB 1v4s 的人葡萄糖激酶的HGPM;下:来在PDB 4no7体系HGPM。 节点根据它们在数据库分子的10% 处的 AUC 值着色。节点越绿,其AUC值越接近1,越红越接近0。“非自然存在的”药效团模型的节点为灰色。从PDB结构获得的药效团模型、出现频次最高的模型以及体系之间具有两个公共特征的节点进行相应地标记。相关的特征向量显示在相应的图上方。
1v4s的HGPM显示了清晰的颜色分离。HF节点区域中的所有节点以及其所有的超集节点都以绿色示出。因此,为了在一致性虚拟筛选性能与模型的数量之间找到最佳平衡,对该区域药效团模型的优先级排序选择可从中获得指示。另一方面,图的上半部分主要描述了性能较差的模型,包括PDB节点。CF节点周围的区域用灰色表示,因为没有对“非自然存在的”节点药效团模型进行虚拟筛选。然而,使用CF节点的所有药效团模型超集的一致性虚拟筛性能良好,尽管这些超集包含性能表现好的和差的单独模型。
4no7 的HGPM是同质的,大多数节点都具有良好的虚拟筛选性能(AUC 大于 0.5)。 HF节点为棕色,其性能低于平均水平。然而,位于图顶部分支的更特异性的节点性能表现良好,包括PDB节点。存在于另一个分支上的自定义选定节点周围的区域也表现良好,这可以解释为不同的配体结合模式。来自与CF节点相同列中的“观察到的”节点的所有模型都显示为红色,因为这些模型的特征数少于三个特征与模型没有明确3D空间叠合,因此不会返回任何命中。
投影到GK蛋白药效团模型分层图的分析
每个单独的体系获得的信息以药效团选择的形式和节点特征组成比较的形式呈现。这是由于药效团特征标记生成算法考虑了配体的哪一部分发生相互作用的,并允许在区分独特特征时有更好的精度。因此,如我们对1v4s和4no7晶体学结构所做的那样,具有不同配体的两个体系之间的比较是不可能的。然而,通过在药效团特征系列产生过程中忽略配体标识符,我们只保留药效团特征和蛋白质标识符相互作用的类型。通过这种方式,我们用配体一侧相互作用发生的准确性来换取在蛋白质一侧“投射”相互作用的能力。因此,这开启了考虑由特定蛋白质产生的所有药效团模型的可能性,而不管哪个配体参与了药效团的相互作用。为了证明这种生成药效团特征序列的替代方法,创建了一个基于1v4s和4no7体系所有的HGPM,如图9所示。关于图的详细信息,见补充表3。
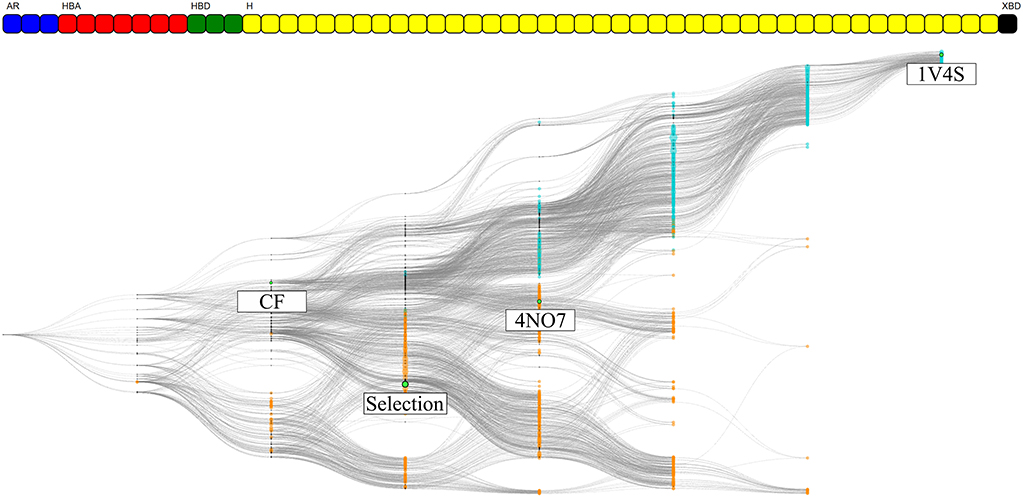
图 9. 从1v4s和4no7体系的MD模拟中获得的HGPM。节点的颜色依据各自来源的体系着色:青色代表1v4s,橙色代表4no7。与PDB结构获得的药效团模型匹配的节点、自定义选择的节点以及具有两个体系公共观测特征的节点被标记并以绿色高亮显示。特征向量显示在图的顶部。
在该分层图中图节点数量和独特药效团特征数量均大于前2个图中的数量,这增加了描述的复杂性。在55个观察到的独特特征中,7个在 1v4s 和 4no7 之间共享:三个氢键受体与精氨酸 63、丝氨酸 64 和苏氨酸 65 相互作用;一个氢键供体与精氨酸 63相互作用;三个疏水特征与酪氨酸 24、异亮氨酸 211、苏氨酸 65 和酪氨酸 214 以及苏氨酸 65 和酪氨酸 214 相互作用。尽管有这些公共的相互作用,但在1v4s和 4no7之间中都没有“观察到的”公共的药效团模型。尽管如此,我们还是可以清楚地观察到两个着色聚类之间的区别,描绘了最初的节点隶属关系。在图的上半部分中青色节点是来自1v4s体系的所有药效团模型,我们可以在其中识别出一个单一分支,其通向PDB结构的专一药效团模型。在图的下半部分,橙色节点代表这来自4no7体系的模型,我们观察到更均匀的间距分布。先前观察到的两个分支不容易区分。可以指出,4no7 PDB 节点位于1v4s 的节点附近,并且自定义选择节点倾向于分叉更多到图的底部,如补充图 6 中更详细地描述的那样。由于特征向量与单个图不同,因此与CF节点具有相同特征的CHA和药效团模型子集的药效团选择与之前不同,该图的虚拟筛选结果细节在补充表4中给出。
基于图的异质性,选择了两个药效团模型。如图10所示,分层图以及两个选定节点,分别是结合模式1(BM1)和2(BM2)。这两种模型都是观察到的最专一的药效团,分别具有7和6个特征。单个公共特征是与精氨酸63相互作用的氢键受体,其对应节点标记为COREF。在图10中药效团模型的3D表示中,我们观察到疏水性和卤键相互作用存在于GK口袋的不同区域,与不同的氨基酸相互作用。由于药效团模型在特征组成与3D叠合方面具有重要差异,以及变构子口袋的存在,观察到的结果可以与不同的结合模式相关联。与两种 PDB 药效团模型之间的初始比较相比,此信息可更好地描述特定结合模式。此外,与CHA或CF+模型选择相比,它允许选择较小的药效团集合用于一致性虚拟筛选。
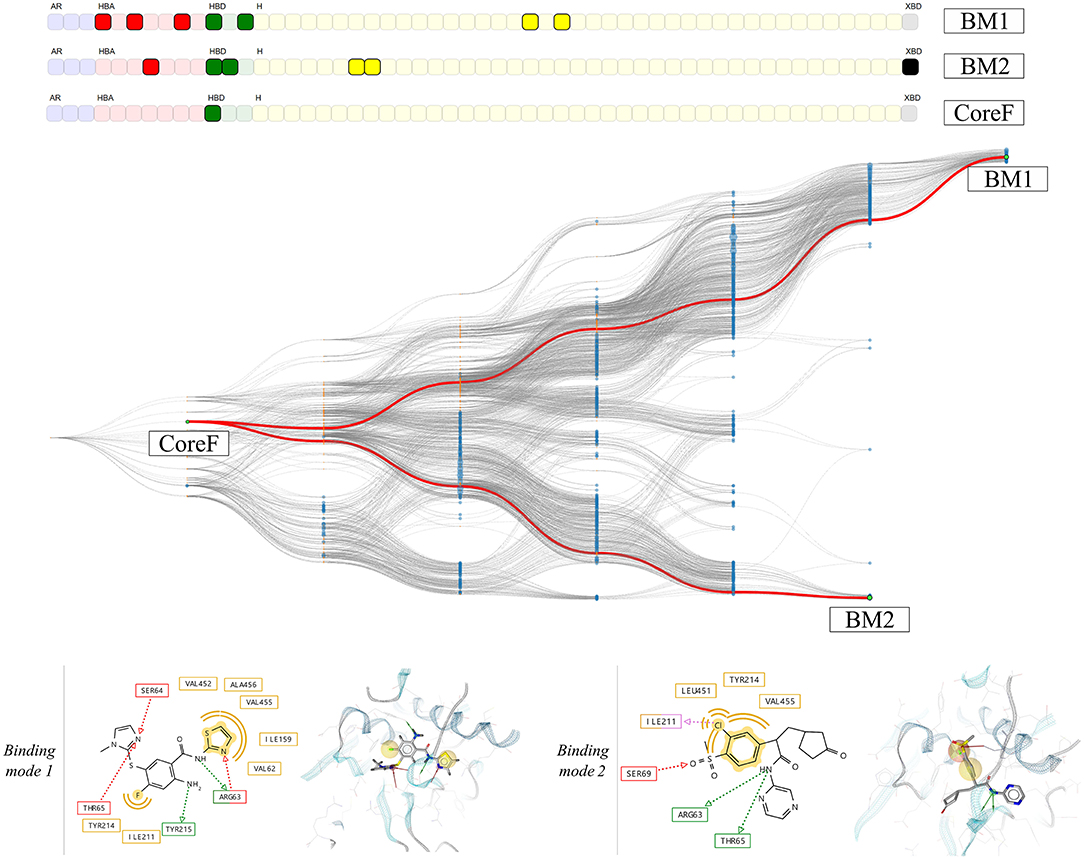
图10. 从1v4s和4no7体系开始进行人葡萄糖激酶MD模拟获得的HGPM。选择节点BM1和BM2作为示例以显示观察到的不同结合模式。对应的药效团模型的2D和3D描述显示在图下面。
结论
出于近期对药效团模型的基于一致性(consensus-based)虚拟筛选方法的兴趣(Wieder et al., 2017; Polishchuk et al., 2019; Madzhidov et al., 2020),我们开发了药效团模型的直观分层图表示。基于药效团图的用户友好的交互式可视化为计算化学家理解蛋白质-配体相互作用模式提供了有价值的信息,并且可以辅助选择用于虚拟筛选实验的药效团模型。该图可以从同一大分子靶的多个晶体学结构或从MD模拟的输出生成的药效团模型的集合来创建,以提供对所研究体系的动态特征的洞察。已经证明图生成在计算上是便宜的,因为即使是对于超过10000个模型的大集合,也只是需要几秒钟的时间(见表1)即可创建。
我们选择了人葡萄糖激酶的两种晶体学结构来评估HGPM生成算法识别不同结合模式的能力,并为一致性虚拟筛选实验选择小的代表性药效团模型集合。对这两个体系分别进行了MD模拟和图生成。使用不同的药效团模型选择来区分这两个研究体系的活性分子和非活性分子。在AUC值和稳定性方面,选择具有最高出现次数的药效团模型中包含的特征超集的所有模型,同时将1v4s体系的模型数量减少20倍,将4no7体系的模型减少45倍以上(见表2),从而有助于显著减少所需的筛选时间。图8中的分层图也有助于通过描述每个药效团模型的虚拟筛选结果来确定性能最好的药效团特征集合。尽管已经注意到4no7体系的分层图(图7)中存在两个专一的分支,但从1v4s和4no7 MD模拟提取的模型(图10)中生成的分层图清楚地识别了GK的两种不同的结合模式,这与文献报道的完全一致(Petit et,2011)。
根据目的的不同,可以通过改变药效团特征串生成算法或通过显示附加属性来灵活地调整图表示。突出了药效团模型之间的分级关系,该图使得用户能够通过在单个图表示中比较几个药效团模型的组成来分析靶标体系,从而促进对结合过程的理解和对药效团模型的选择以用于一致性虚拟筛选。使用由图表示提供的信息的典型工作流,首先选择具有最高观察频次的单个药效团模型,然后执行虚拟筛选,并且最后添加或移除通过遵循分级链接而识别的个体特征,以构建具有准确度和特异性之间的最佳比率的精细模型。药效团模型分层图的提出,我们希望引入多个药效团模型的直观表示,并为计算和药物化学家提供一个新的工具,使他们能够更好地理解蛋白质-配体结合过程,从而在优化生物活性分子的过程中提供更好的决策支持。
数据获取声明
本研究中提供的数据集可以从在线存储库中找到。存储库的名称和登录号可以在以下位置找到:http://www.wwpdb.org/,1v4s http://www.wwpdb.org,4no7。
补充材料
本文的补充材料可在线获取,见:https://www.frontiersin.org/articles/10.3389/fmolb.2020.599059/full#supplementary-material
文献
略
HGPM演示
URL:https://www.inteligand.com/hgpharm/demo
相关主题
分子动力学模拟的轨迹现在可以大规模产生,然而却难以解释并用于指导设计、优化先导化合物。Thierry教授在他的一个报告中讲述了如何用药效团技术结合分子动力学模拟指导先导化合物优化:https://www.bilibili.com/video/BV1Xy4y1r791


