
用分子动力学模拟生成蛋白构象系综用于对接实验更具生物学合理性
摘要:对接是一种利用蛋白结构产生配体结合模式(pose)并对所产生的相互作用进行打分的方法。在药物发现中,该方法广泛用于筛选分子并进行优先级排序。对接软件通常将蛋白视为刚体,而配体的构象是柔性的。有时这种处理方式是有问题的,因为对于一个给定的配体,蛋白结构并不是在生物学上合理的构象,这导致对接结果与生物学不相关。 在本文中,我们首次在Flare里验证了使用分子动力学(MD)模拟来产生蛋白构象系综以用于系综对接的方法。
Flare™蛋白分子动力学模拟用于构象系综对接
作者:Ryuichiro Hara†, Stuart Firth-Clarke†, Nathan Kidley†
单位:†Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK
编译:肖高铿/2022-10-25
前言
X-衍射蛋白结构可能不具有生物学意义构象的原因有几个,例如,晶格内相互作用产生的晶体堆积伪影、纯化或结晶过程中使用的辅因子(cofactor),或者蛋白结构是在其活性位点里没有配体存在的情况下解释得到的。此外,晶体结构代表了一种时间的快照,并不能反映蛋白的动态性质。当对接将蛋白质视为刚体时,不考虑蛋白的柔性可能会导致错误的结果。蛋白的柔性可能意味着氨基酸侧链的微小移动,从而导致蛋白结构的较大变化。蛋白构象变化影响刚性对接的一个例子来自激酶,取决于抑制剂的结合与否可观察到蛋白的DFG-loop至少有两种构象DFG-In和DFG-Out。不考虑这一变化会产生与化合物活性无关的对接结果。
Cambell等人1报道了如何利用分子动力学模拟对蛋白的柔性进行采样并对轨迹进行聚类以生成蛋白构象系综。然后将配体对接到蛋白构象系综之中,其中每个蛋白构象都作为刚性处理,这样间接地将蛋白柔性纳入到对接计算中。以CDK2和FXa为例进行了对接和交叉对接实验,结果表明该方法能够为给定的配体在打分值最低时得到正确的结合模式(编著注:打分越低代表结合亲和力越强)。
CDK2与FXa数据集1也用来验证我们的方法。在两个算例中,用Flare对X-衍射蛋白结构进行了结构准备与分子动力学模拟。对模拟轨迹进行聚类以产生不同蛋白构象的系综,然后将其用于Flare的系综对接(Ensemble docking)3。分子动力学模拟轨迹聚类是Flare即将推出的一项新功能。该工作流程可以为对接的每个配体找到最佳的蛋白构象,从而能够将蛋白的柔性纳入对接工作流中。
Flare除了具有友好的用户界面之外,还有一个Python API,使得用户能够通过Cresset Engine Broker™的隐式分布式计算特性以编程方式自动化地运行工作流。在我们的原型工作流中,我们开发了Python脚本来完成这项研究。
方法
蛋白的准备
所有的蛋白结构都用Flare中的蛋白准备工具来进行准备。下载结构并检查氨基酸侧链的替代构象,使用侧链的占据率或基于侧链的接触优选的侧链旋转异构体。这些操作是使用Flare的自动化蛋白准备工具来完成。该工作流的操作包含了:添加氢原子,探索可解离残基的互变异构体状态,添加缺失的侧链,并填补结构中两个氨基酸之间的缺口,末端残基的封端,优化残基侧链以遍最大化相互作用和内部氢键网络。在大多数情况下,Flare中的自动蛋白质准备适合于对接、能量计算与分子动力学模拟等研究,但仍建议对准备结果进行仔细地检查。
分子动力学模拟
保留配体周围的结晶水分子并用于Flare MD模拟。大多数MD参数使用Flare的默认值,其中小分子加AM1-BCC电荷并用OpenFF2.0参数化。蛋白加Amber14ffsb电荷并参数化。氢质量重分配(hydrogen-mass repartitioning,HMR)使用4fs的时间步长。在100ps平衡后进行4ns的模拟。这使得每个轨迹产生2000帧快照(每帧间隔2ps)。使用默认设置,用户只需在Flare应用程序中点击几下即可运行MD,并且在配备有一个GPU的PC上不到一小时即可完成4ns的模拟。
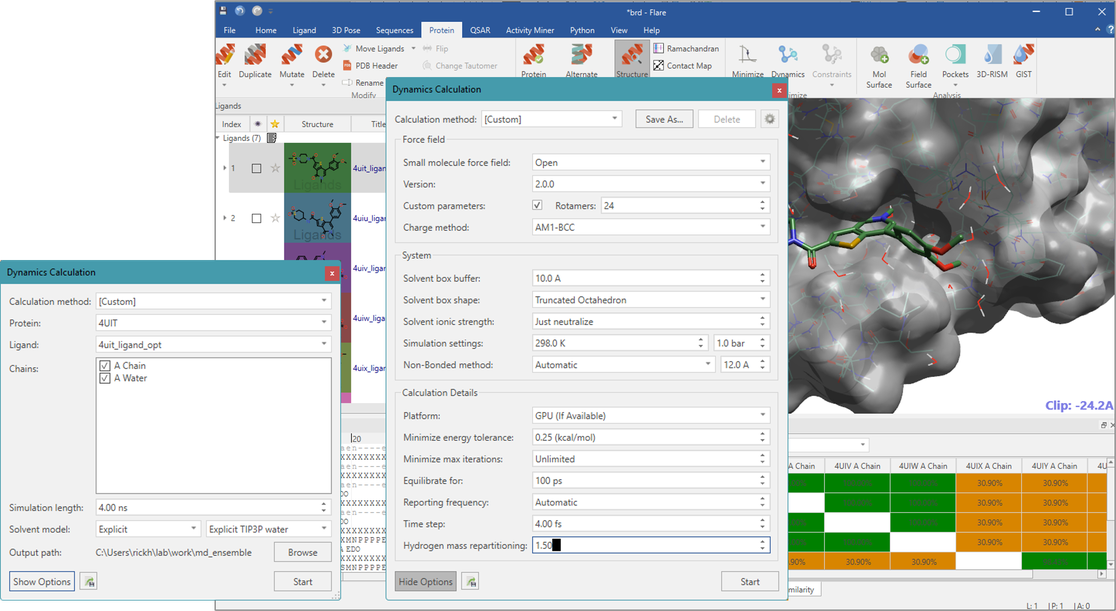
图1. 在Flare中创建MD作业
轨迹聚类
活性位点被定义为在共晶体结构中距离配体6Å内的一组残基。活性位点残基的重原子的坐标向量用于基于质量加权平均连接RMSD的聚类算法1,2。Cambell等人提到,6-8个簇(cluster)足以构成一个系综,但这里我们从轨迹中采集20个代表性构象生成20个簇。虽然可以使用AmberTools的cpptraj函数来完成轨迹聚类,但我们选择在Python中实现聚类,以探索不同的选项并优化聚类结果。
图2描述了簇之间的时间依赖性转换,RMSD跟踪了活性中心中重原子的波动。图2中的着色是根据簇成员关系而定,图节点的大小对应于频率,有向边的长度对应于转换的频率(越短越频繁)。 这呈现了分子动力学模拟的蛋白结构空间循环和进程(尽管为了清楚起见省略了非常频繁的自循环)。虽然平均连接并不平均地划分簇的大小,但是计算了每个簇内基于RMSD的方差以识别中心点(medoid),而不论簇的大小。中心点构象都是来自轨迹的某些快照,因此最好使用与MD过程相同的力场来对之进行能量最小化。由于我们只对活性位点周围的直接环境感兴趣,因此对于每个快照结构,距离配体8Å以内的原子进行了能量最小化。
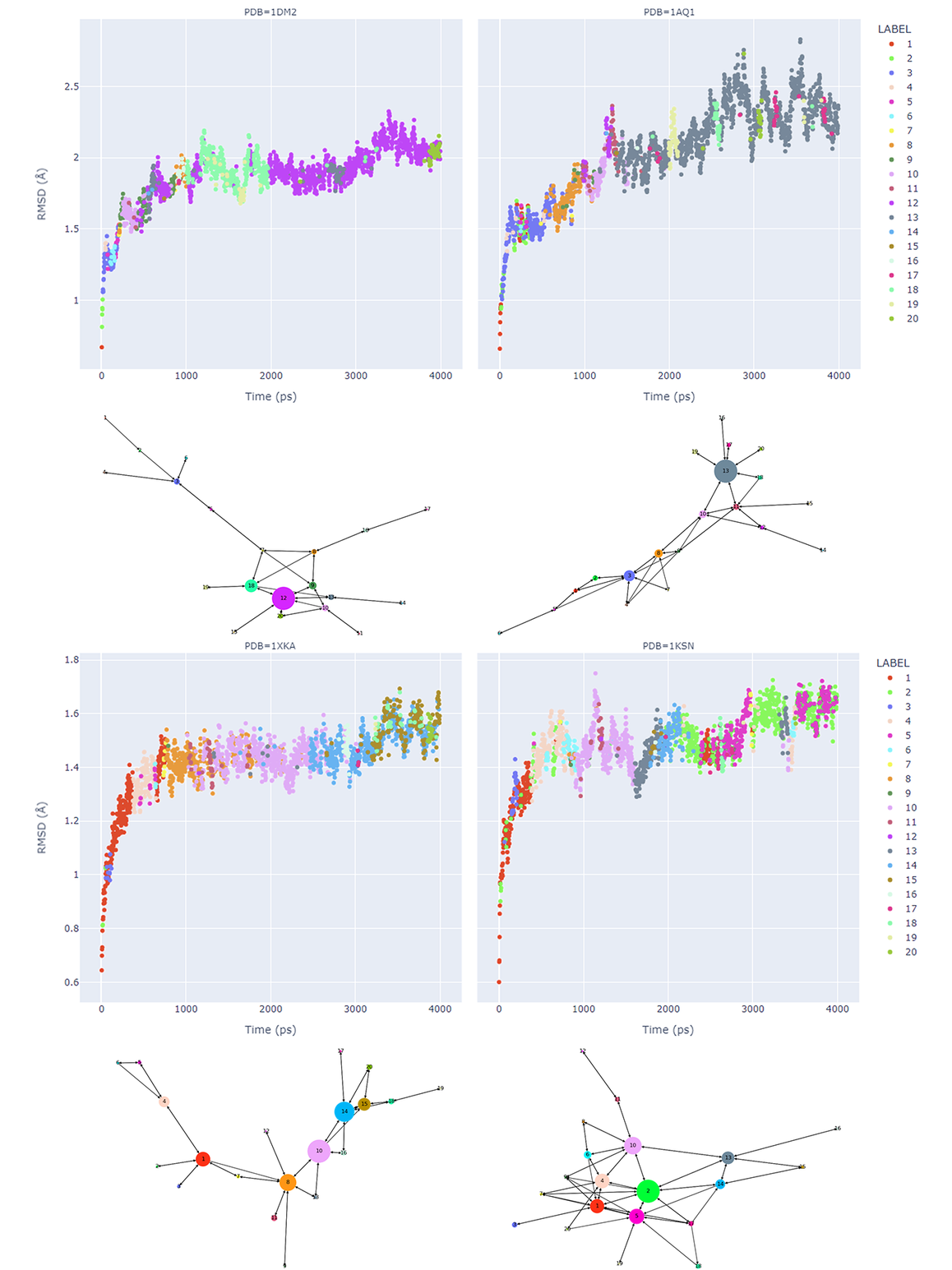
图2. 动力学轨迹分析,对于每个蛋白质结构1DM2、1AQ1、1XKA、1KSN,显示了与起始坐标相比的RMSD值。 然后对轨迹进行聚类,结果显示在有向图中。每个示例之间的簇节点和簇成员的着色是一致的。节点大小对应于簇成员的数量,边的长度对应于转换的频率(越短越频繁)。 这显示了在动力学模拟过程中蛋白结构空间的循环和进程(尽管为清楚起见省略了极其频繁的自循环)。
对接计算
Flare系综对接工作流允许您将化合物对接到同一蛋白的多个构象里。 我们使用Flare Python API编写pyflare脚本来执行对接实验,调整GUI中所有的实验设置,例如对所有的蛋白添加统一的约束,并将结果保存为自定义的格式。整个系综加上X-衍射结构的所有高分结合模式都保存到SDF文件中。这使用户能够使用其他外部工具(如Jupyter笔记本等)进行快速地分析。
Flare内置的Lead Finder™算法3用于所有对接计算。每个选定的蛋白构象(簇中心点和参比X-衍射结构)对一组配体的对接计算迭代进行。对于每个对接过程,都会生成可能的配体结合模式(pose),并用各种分值表示其与结合位点的拟合质量。虽然Lead Finder提供不同的基于能量的打分(dG、VSscore和Rank Score),但我们使用经过优化的Rank Score来识别结晶的配体结合模式,用dG打分来比较配体自身对接和交叉对接实验计算产生的结合模式。
本文遵循的步骤总结在图3中。黄色显示的操作步骤在Flare中执行;聚类步骤用Python编写并pyflare API来执行。

图3. Flare/pyFlare的MD与系综对接流程
结果
分子动力学模拟系综对接的验证
正如Cambell论文中所解释的那样,用两个配体数据集的自身对接(self-docking)和交叉对接(cross-docking)验证了该流程。这两个数据集分别是:来自PDB结构 1DM2与1AQ1的两个 CDK2 配体(图 4 – 左);以及来自PDB结构1XKA和1KSN的两个FXa 配体(图 4 – 右)。 CDK2 配体提供了结合配体具有大尺寸差异的算例;FXa配体是具有高度柔性的算例。在这两种情况下,交叉对接可能经常会失败。

图4. MD/系综对接的验证。左:CDK2 配体的自身对接和交叉对接结果。 右:FXa配体的自对接和交叉对接结果。
对于CDK2算例(图 4 – 左),共晶配体HMD(来自PDB 1DM2)与STU(来自PDB 1AQ1)对X-衍射蛋白结构的自身对接RMSD均在1Å之内。HMD与PDB 1AQ1的交叉对接相当成功,RMSD = 1.554 Å。然而,由于配体的大尺寸和由此产生的口袋尺寸差异,STU与PDB 1DM2的交叉对接未能产生正确的结合模式。
正如预期的那样,CDK2(HMD 和 STU)针对来自其各自蛋白结构的MD快照系综的自身对接实验也产生了良好的结果。与之前的直接交叉对接实验不同,使用MD快照系综进行的交叉对接实验都是成功的。与原始的X-衍射共晶配体相比,对接结合模式的RMSD均小于2 Å(STU 的RMSD = 1.255 Å ,HMD的RMSD = 1.654 Å)。值得注意的是,除了这种可接受的RMSD要求外,还对对接结合模式进行了可视化检查,确认它们与共晶结合模式相似。
具有高度柔性的FXa配体则是更困难的算例,配体4PP(来自PDB 1XKA)与FXV(来自PDB 1KSN)的自身对接是成功的(图 4 – 右)。但交叉对接实验没有那么成功,对于对接得分较差的两个配体所产生结合模式的RMSD均大于2 Å。对这些结合模式进行目视检查证实,结果里不包含任何的共晶结合模式。
4PP和FXV配体针对来自其各自蛋白结构的MD快照系综的自身对接实验以及针对晶体蛋白的刚性对接实验得到略微改善的RMSD值。FXa配体的针对MD快照系综交叉对接实验产生的结合模式其RMSD在可接受的范围内(FXV的RMSD =1.385,4PP的RMSD = 1.498),LF dG打分值接近自身对接实验中观察到的值。
总体而言,CDK2和FXa算例的结果表明,Flare中的MD-聚类-系综对接方法可以成功地产生蛋白构象的系综,从而能够将蛋白的柔性纳入到对接工作流中。在这些算例中,活性位点的差异主要在于活性位点的侧链运动,因此用的相对较短的4ns MD模拟足以采集到此类变化。蛋白构象的更大变化可能需要更长时间的MD模拟来生成适合系综对接的构象系综
结论
在本文中,我们提出了一种方法,它利用Flare的系综对接工作流与分子动力学模拟一起进行对接实验,同时考虑到了蛋白的柔性。当可用的蛋白结构不是您配体合适的生物学相关构象时,该方法特别有用,这种情况还可能体现为氨基酸的侧链运动或更大的构象变化。
此处使用的分子动力学模拟聚类方法还在开发中,在即将发布的Flare 中实现。它是MD模拟的一种有用的分析方法,因为它能够生成不同的蛋白构象,以间接地方式考虑了对接实验中蛋白的柔性。
在本文中,使用Python脚本来自动化地进行蛋白准备、系综对接和结果分析等整个过程。这些脚本可在Jupyter笔记本上进行保存与执行。
文献
- Campbell, A. J.; Lamb, M. L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. J. Chem. Inf. Model. 2014, 54 (7), 2127–2138. https://doi.org/10.1021/ci400729j.
- Shao, J., W. Tanner, S., Thompson, N., & E. Cheatham, T. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. Journal of Chemical Theory and Computation, 2007, 3(6), 2312–2334. https://doi.org/10.1021/ct700119m
- Lead Finder, 2112 build 1, BioMolTech®, Toronto, Ontario, Canada


