
摘要:本文介绍了常见偏电荷(partial charge)的计算方法、区别与合适的应用场景。OpenEye的QUACPAC提供了多种偏电荷计算方法,尤其是ELF构象选择规范的AM1-BCC偏电荷计算特别适合于分子动力学模拟、FEP计算等涉及配体构象可能发生较大改变时的配体偏电荷计算,以确保您获得最佳计算性能与可靠性。
前言
为了从静电计算中获得有意义的结果,对小分子配体和生物大分子(如蛋白质和核酸)分配适当的原子偏电荷(partial charge)至关重要。
分子可以被认为是原子核及其周围电子的集合。每个原子核中质子的数量决定了它的原子序数/元素(atomic number/element)。如果电子的数量与原子核中质子的数量完全匹配,则分子是中性的,没有净电荷。如果电子多于质子,则分子具有净负电荷,如果电子少于质子,则该分子具有净正电荷。
定义分子身份的是原子核和净电荷。事实上,这是量子化学中常见的表示。从分子中添加或移除掉一个电子(或原子)会产生不同的分子。
在化学信息学的离散世界中,价键理论允许系统中存在的电子以具有规范键级(formal bond orders)的键和分配给特定原子的形式电荷(formal charges)来表示。形式电荷的总和等于分子上的净电荷(net charge),但由于共振离域,哪些原子被分配了哪些形式电荷在某种程度上是任意的。在这种情况下,同一分子可以用类似的连接表(connection table)来表示,但形式电荷分配给不同的原子集。
例如,胍可以表示为具有分配给碳形式电荷的N[C+](N)N,或者表示为[NH2+]=C(N)N,具有任意分配给其他等效氮之一的形式电荷。类似的例子还有硫代羧酸基团,其中C(=O)[S-]或C(=S)[O-]都是相同化学官能团的同样合适的表示。
两性离子(zwitterion)是一种电中性分子,表示为同时含有正形式电荷原子和和负形式电荷原子的分子。
当我们讨论原子上的形式电荷时,也许最重要的事实是,它们都是化学家为适应特定的化学模型而人工构建的,是化学家们根据其狂热的想象而虚构出来的产物。就像价键理论一样,它们是一个非常有用、强大的宇宙离散模型。但与任何现实模型一样,它也有其局限性。尽管形式电荷对人类有诸多好处,不幸的是,但它们并没有局限于原子上。
即使在化学信息学中,用价键理论描述形式电荷的局限性也是显而易见的。例如,Sydnone是一类杂环化合物,如果不引入并任意分配的正电荷和负电荷,就无法使用正常的共价键来画出结构。类似地,在无机化学中,二锝阳离子Te2+5会引起类似的问题,即+5形式电荷不能在不破坏对称性的情况下分配给两个锝原子。
描述分子周围电子密度分布的波函数(wave function)的一个更好的模型或近似是使用原子偏电荷(partial charge)。偏电荷是分配到每个原子中心的浮点值,用于模拟分子上电子的分布。
原子偏电荷是另一种近似,很像上述的形式电荷。然而,偏电荷提供了一个更好的模型来描述分子的电场、偶极矩和其他可观测性质。
使用偏电荷的一个常见局限是假设它们是不随着分子构象变化而变化的。不幸的是,分子周围电子的分布取决于其原子核的空间构型。一些偏电荷分配算法,如Goddard和Rappé的方法,考虑了这些构象效应;而其他基于量子力学的算法,如Bayly等人提出的RESP和AM1BCC方法,则竭尽全力消除构象效应,例如通过限制和对称化对称原子位置。为了能够用一组原子电荷正确地处理多个构象和几何结构的变化(例如几何结构优化),消除构象效应是非常必要的。
QUACPAC的电荷计算方法
Marsili-Gasteiger偏电荷
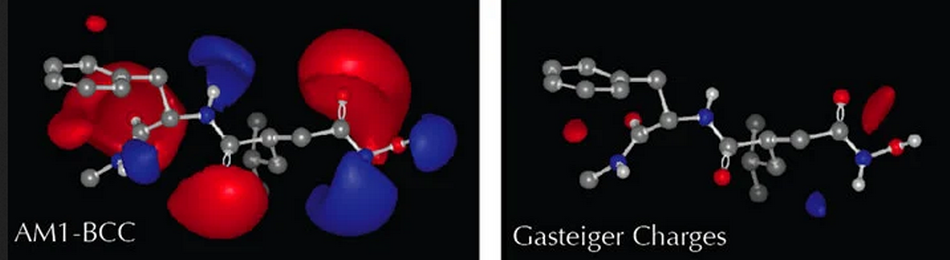
Marsili-Gasteiger偏电荷分两步法计算。第一步,给分子中的每个原子分配种子电荷。例如,每个羧酸根氧分配的值为-0.5。第二步,这些初始电荷通过键共享,将一定量的电荷从一个原子移动到另一个原子。移动的那部分电荷及其方向由键两端原子的电负性差决定。然后用松弛算法(relaxation algorithm)迭代几次(默认为八次),每次迭代移动的电荷都将衰减。OpenEye不建议将此电荷模型用于分子间相互作用;它从来就不是为了这个目的。该方法的作者(Johann Gasteiger)开发该方法目的是比较不同分子背景下相关有机官能团的相对反应性。本文提到Marsili-Gasterge电荷仅是为了与其它电荷模型进行比较。
MMFF94偏电荷
MMFF94和MMFF94s力场所使用的偏电荷分四步分配。第一步,为分子中的每个原子被分配一个MMFF94原子类型。第二步,根据每个原子的原子类型为其分配初始的种子偏电荷。对于少数原子类型,初始偏电荷也取决于局部环境。第三步,将分配到芳香环的初始电荷与所在芳香环的所有原子之间共享。最后,第四步,根据键的键类型(单、双、三)以及键两端原子的原子类型,根据键电荷增量表(bond charge increments,BCI)在键之间移动电荷。它们是为上述力场内的静电相互作用而开发的,是与这些力场一起使用的合适电荷,尤其是用于药物和生物有机小分子的分子内相互作用。它们不太适合(但仍然可以通过)用在Amber、Charmm和Gromacs中采用两体加和库仑相互作用法来计算常见的分子间相互作用。对于此类力场有更好的选择,蛋白质和肽使用amber99sb电荷,配体使用am1bccsym电荷。
AM1电荷
AM1电荷是从半经验量子力学计算中导出的一套Mulliken类型电荷。有关该方法的进一步讨论,请参见Dewar(1985)的文章。这类电荷不可应用于力场来计算分子间相互作用。
AM1BCC电荷
AM1BCC电荷是从AM1半经验量子力学(QM)波函数衍生出的Mulliken类型偏电荷。对每个原子上的偏电荷进行键电荷校正(bond-charge correction,BCCs),以产生新的偏电荷。因为几个不同因素对这类QM衍生电荷产生显著的影响,OpenEye的QUACPAC提供了多种不同的AM1BCC电荷变体。具体来说,这些影响因素包括:
- 优化:输入的几何结构是否得到优化。
- 对称性操作:拓扑相似的原子(例如羧酸根上的两个氧)是否被限制为具有相同的值。
QM波函数通常对几何结构非常敏感,尤其是键长和键角,因此这会显著影响偏电荷。通常,建议进行几何优化。为了避免由于强烈的分子内静电相互作用而导致构象崩溃,对起始的几何结构施加了轻微的约束。
真正的QM波函数通常在拓扑相似的原子周围是不对称的,导致不对称的偏电荷。然而,如果在不同的构象上使用相同的偏电荷(比如普通的固定电荷力场),重要的是这些电荷是对称的,或者在形式上简并的构象之间相互转换(例如羧酸根的180度旋转)将产生非简并的静电能。一般来说,如果偏电荷想要应用于多个构象上,强烈建议使用对称性操作。
AM1BCC电荷的另一个重要问题是,如果产生了不适用于其他构象的高度构象特异性电荷,则会导致那些其他构象体产生不希望的静电能扰动。为了解决这个问题,我们强烈推荐下面描述的ELF构象选择规范。
“标准”的AM1BCC既包括优化,也包括对称化操作。
OpenEye认为AM1BCC电荷是目前可用的最佳偏电荷模型。如需进一步讨论,请参阅Christopher I. Bayly的著作。
ELF构象选择规范
ELF构象选择是一种从大型构象异构体数据库中选择一个或多个具有静电最小相互作用官能团(Electrostatically Least-interacting Functional groups,ELF)的构象异构体方法。该方法的目的是解决一般QM衍生电荷的重要问题,包括AM1BCC电荷。问题是,特定构象专一的强短程分子内极化,如分子内氢键或盐桥,通常会给所涉及原子的偏电荷带来强烈扰动。这些电荷与不具有分子内极性相互作用的其他构象的电荷非常不同。如果将这样的偏电荷应用到所有构象上,那么其他构象异构体很可能具有错误地过度稳定的溶剂化能。
由此问题引起的第二个问题是由QM衍生的电荷产生的静电能的精度或灵敏度。我们的意思是:不同构象之间的静电相对能量的变化取决于QM衍生电荷使用的构象。想象一个分子有两组不同的QM衍生偏电荷,每一组都来自具有不同强分子内氢键的构象。对于发生分子内氢键相互作用的原子,每组偏电荷都对其有强烈的扰动,但会有所不同。构象之间的相对能量将根据所使用偏电荷的不同而不同。
由于这些原因,重要的是需要避免从具有静电强相互作用官能团的构象产生QM衍生偏电荷。这就是ELF构象选择规范的作用。
ELF需要从足够多的构象异构体开始,这样它才能找到一个不具有强相互作用官能团的构象异构体集合。第一步,使用MMFF94偏电荷绝对值(用绝对值替换原始的负电荷)来计算每个构象的库仑静电能。此电荷的静电能会使所有的强极性相互作用不稳定,从而使得最低的静电能对应于静电相互作用最少的官能团。选择能量最低的2%构象作为2%ELF构象集。我们发现,即使对于高极性和带电的分子,平均从2% ELF构象集中取10个多样的构象也足以找到一组具有良好的QM衍生偏电荷。
Amber ff94、ff96、ff99、ff99sb与ff99sbc0偏电荷
AmberFF94力场所使用的偏电荷是基于拟合量子力学静电势(ESP)。它们的开发是为了解决早期静点势拟合电荷的两个关键问题:在电荷中心上不切实际的高电荷以及原子电荷随构象而变化。虽然后者在电子结构方面应该有一定的基础,但电荷拟合过程中的数值不稳定性是这两个问题的根源。AmberFF94电荷使用约束的静电势拟合(restrained esp-fitting,RESP)来控制数值不稳定性,同时使用多构象拟合来产生仅限于单个残基的构象无关电荷。要特别注意确保主链酰胺具有一致的电荷。AMBER力场ff94、ff96、ff99、ff99sb和ff99sbc0都使用相同的RESP电荷,而在其他作用项上具有差异。


