
摘要:Cbl-b抑制剂与Tyr363相互作用的TEG片段长期以来都保留了1,2,4-三氮唑结构,直到阿斯利康的Quinn等人采用生成式AI与SBDD相结合,经过多轮的DMTA循环最后发现新型TEG——六员环的胺基甲酸酯(Carbamate)。在本文中,我们采用Spark基于配体相似性打分对1,2,4-三氮唑进行生物等排体替换,一步计算就从结果里以排名第2的方式找到了六员环的胺基甲酸酯,重现了阿斯利康Quinn等人化合物12的TEG设计。这使得DMTA循环次数减少,可以加速胺基甲酸酯 Cbl-b 抑制剂的发现。这说明了,即使生成式AI与人类专家都难以实现的跃迁,Spark依然可以提供操作简单、结果可靠、高效的生物等排体替换方案。
肖高铿/2025-01-22
前言
Casitas B淋巴瘤原癌基因-b (Cbl-b) 是一种RING指型E3连接酶Cbl家族成员1,并且作为免疫细胞受体信号通路的负调节因子,在癌症免疫治疗中扮演着重要角色2。由于T细胞激活的抑制会建立一个免疫抑制的肿瘤环境,因此Cbl-b成为癌症治疗中的一个有吸引力的目标3。
Cbl-b的催化位点位于其N端,包括酪氨酸激酶结合(tyrosine kinase binding,TKB)域,该域负责招募底物;连接连接臂螺旋区(linker helix region,LHR),以及一个负责E2~泛素招募的RING指域。LHR包含一个在Cbl蛋白中严格保守的酪氨酸残基,对功能至关重要,因为这个酪氨酸的磷酸化(Cbl-b中的Tyr363)会促使蛋白质进入活性状态。因此,通过小分子与Tyr363的相互作用应该可以抑制Cbl-b的整体功能。
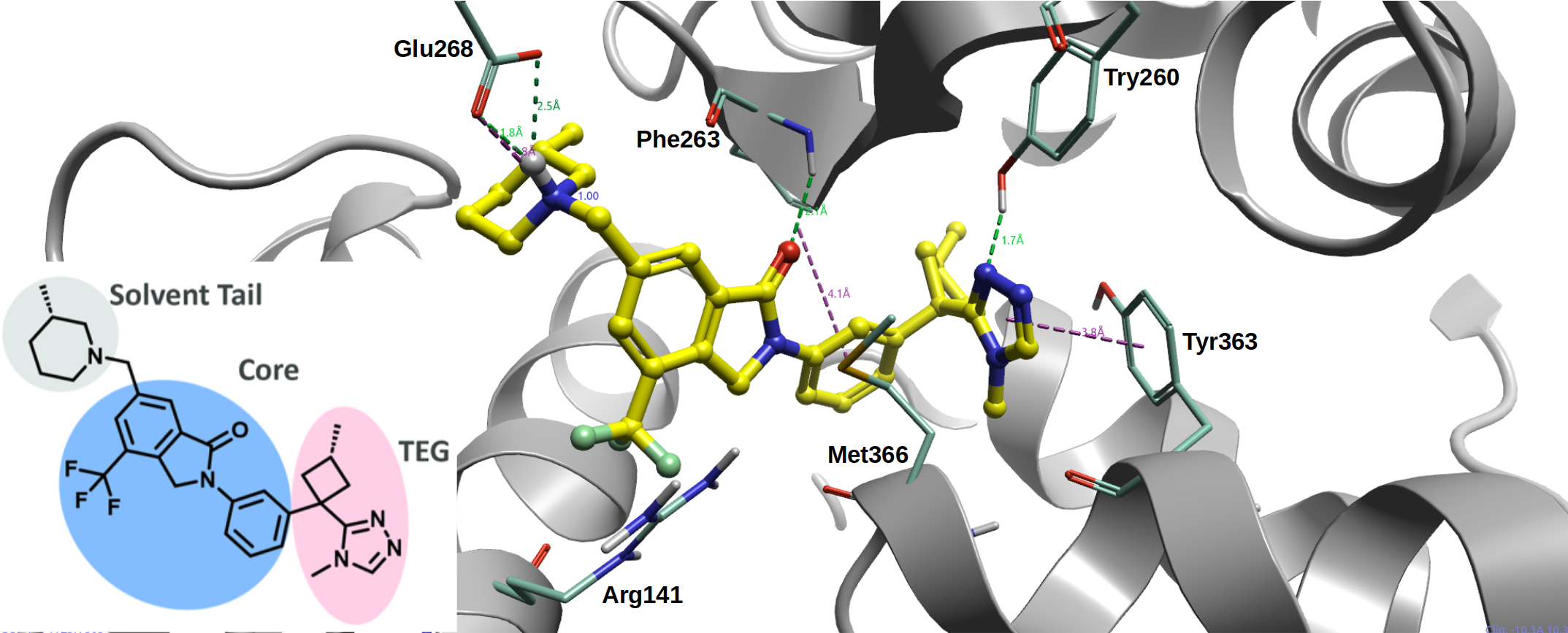
图1. Cbl-b抑制剂NX-1607与Cbl-b的共晶体结构(PDB 8GCY),突出显示了配体与结合位点的关键相互作用。2D结构是Cbl-b抑制剂NX-1607,分为三个部分:蓝色表示的母核部分,灰色表示的溶剂尾部,以及粉色表示的酪氨酸结合基团(Tyrosine Engaging Group,TEG)。
Barsanti等人4公开了一系列以NX-1607(图1)为代表的N-芳基异吲哚酮类Cbl-b抑制剂,这些化合物来源于高通量筛选活动中的一个苗头化合物5,6。设计重点在于保持抑制剂的分子内“胶水”功能,通过与Tyr363的相互作用,阻止Cbl-b进入其LHR和RING域的开放状态,这两个区域负责结合E2~泛素。最近发布的NX-1607晶体结构(PDB 8GCY)展示了这类化合物的结合模式(图1)。异吲哚酮核心部分作为氢键受体与Phe263的主链相互作用,并作为C-H氢键供体参与互动;而暴露于溶剂的哌啶环则与Glu268的侧链相互作用。1,2,4-三氮唑部分与蛋白质发生了三个重要的相互作用:与Tyr260的酚基形成氢键、与催化相关的Tyr363进行π-π堆积,并与Tyr363的主链羰基氧形成C-H氢键。由于Tyr363不能再与溶剂接触并且与抑制剂形成了新的相互作用,因此对于功能所必需的磷酸化过程被阻止了。
除了Nurix团队的研究之外,其他团队的研究也报告了通过与Tyr363结合来抑制Cbl-b的情况,这些抑制剂的共同点是都包含了1,2,4-三氮唑这个TEG片段。如图1所示,这些抑制剂由三个部分组成:核心部分(core)、溶剂尾部(solvent tail),以及与Tyr363相互作用的TEG片段(图1)。没有新型TEG片段被文献报道,这也间接说明了三氮唑TEG片段的生物等排体替换是相当难的。
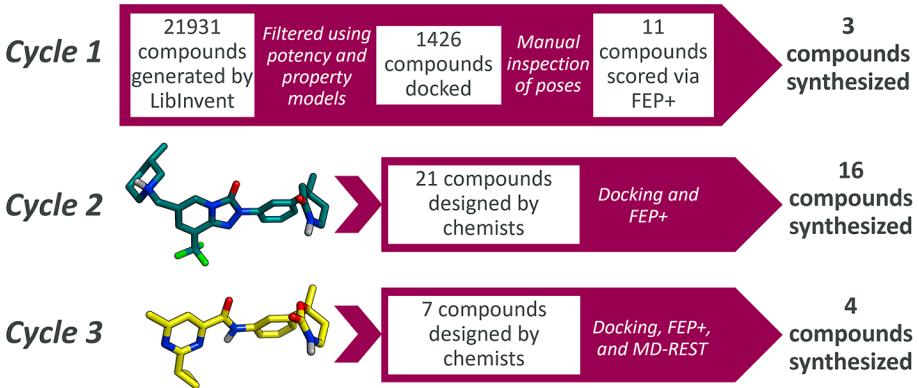
图2. 三轮DMTA循环过程的总结,其中在第一轮循环中,筛选标准包括ROCS打分、预测的HLM CLint、预测的Log D值、氢键供体数量、化合物电荷以及总的MPO(多参数优化)打分。
为了寻找Cbl-b抑制剂,最近阿斯利康的Quinn等人7报道了结合了生成式人工智能设计引擎REINVENT与基于结构的药物化学设计,以发现新的Cbl-b抑制剂。这一努力成功的关键在于“设计”阶段的进化,即在Design-Make-Test-Analyze循环中的设计阶段引入了多轮迭代的计算机辅助结构基础药物设计(图2),并在选择合成之前,由基于物理的亲和力预测和机器学习的DMPK(药物代谢动力学和药代动力学)预测模型进行指导。这加速发现了一系列强效的含氨基甲酸酯(carbamate,图3)TEG片段的Cbl-b抑制剂。
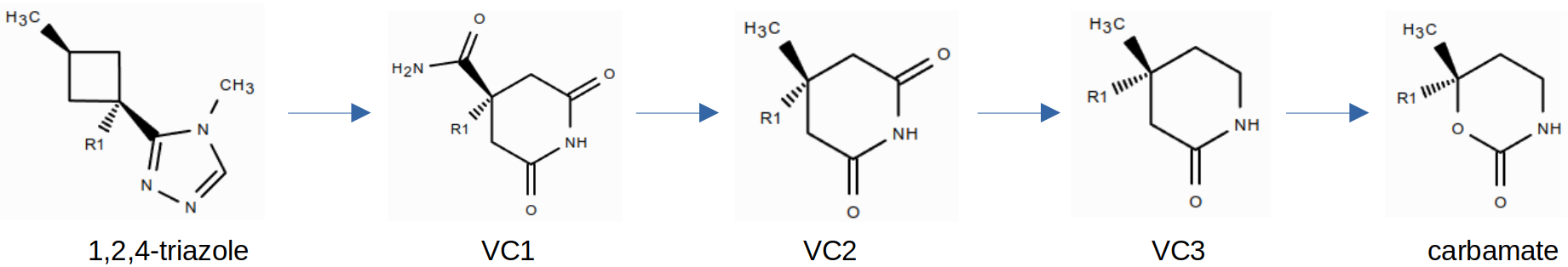
图3. TEG片段从1,2,4-三氮唑约跃迁为氨基甲酸酯的设计过程
然而,TEG片段从NX-1607的1,2,4-三氮唑跃迁到6员环氨基甲酸酯(carbamate)的设计过程并非通过REINVENT的AI生成一步完成,而是在AI生成的基础上通过药化专家多轮基于结构的优化设计得到,见图2、3与4。
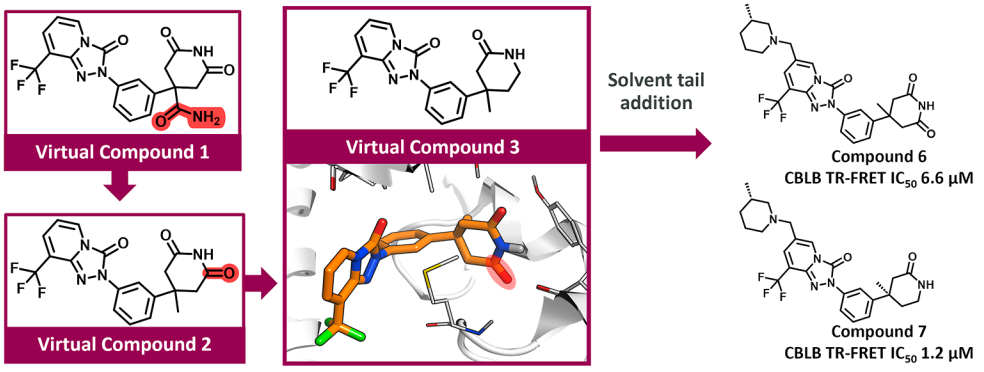
图4. REINVENT的AI生成结果及基于项目后续SAR设计的虚拟化合物,从中合成了化合物6和7
首先,在第一轮的DMTA循环中(图2),作者从REINVENT生成的21931个结果中,通过性质过滤、分子对接、FEP评估结合亲和力等等,选中了虚拟化合物1(Virtual Compound 1, VC1,图4)。此化合物激发了由药物化学驱动的设计,通过计算结构分析进行了筛选,包括对接和自由能微扰计算(分别使用Glide和FEP+执行)以及基于性质的机器学习模型对Log D和HLM内在清除率进行排序。对接结果显示,酰亚胺VC1将一个胺甲酰基置于一个之前只容忍疏水基团的季碳上,这导致设计出了化合物VC2(图4)。两个虚拟化合物都进行了FEP+分析,预测VC2相比VC1具有显著改进的结合自由能(约3 kcal/mol)。对接模型的进一步结构分析表明,酰亚胺上的一个羰基可能不会形成显著有益的额外相互作用,实际上可能会与附近的主链羰基产生不利的电荷-电荷排斥(图4),这导致了内酰胺类似物VC3的设计。在合成前,酰亚胺和内酰胺设计均通过引入溶剂尾部的哌啶基团进行了进一步优化,最终合成了6和7(图4)。化合物6、7在生化测定中分别显示了6.6和1.2 μM的IC50抑制活性。尽管活性不是很强,但考虑到它们的TEG化学类型在结构上与已知的1,2,4-三氮唑不同,这一结果仍然令人鼓舞。重要的是,获得了活性异构体7的晶体结构,确认了内酰胺季碳上的R型立体化学以及分子的结合模式。
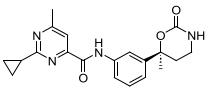
图5. 氨基甲酸酯化合物12
7与Cbl-b的共晶结构(PDB 9FQI)表明,7的内酰胺环为了保持与Tyr363和Tyr260的两个氢键相互作用而采取了高张力能构象。因此第二轮的DMTA循环的目标是在保持氢键相互作用的同时减少这种构象张力能,从而改善结合亲和力。药物化学设计集中在向环中引入杂原子并改变杂环的环大小。如前所述,所有设计均使用FEP+和机器学习模型对HLM CLint和Log D进行了评估。经过数轮的迭代设计后,共选择了12个化合物进行合成,其中六元环的氨基甲酸酯衍生物12(图5)的结合亲和力为4.8 μM,人肝微粒体清除率为5.9 μL/min/mg。
在成功识别出氨基甲酸酯12之后,第三轮的DMTA循环专注于通过增加从手性中心碳延伸出的位阻体积来进一步改善结合亲和力。在证明了新型环丙基氨基甲酸酯TEG的实用性后,该片段还与其他强活性的母核结构进行组合探索。将新的氨基甲酸酯TEG与项目平行研究努力中识别的吡啶酮母核组合,得到了强效的类似物24(未显示结构),其脂溶性降低,并且体外清除率进一步改善。这些例子突显了环丙基氨基甲酸酯基团可以与多个Cbl-b抑制剂母核结合使用,提供具有适当性质的高活性分子,以便进一步优化先导化合物。
本文将描述如何用SPARK8一步实现从NX-1607的三氮唑TEG跃迁到氨基甲酸酯(carbamate)TEG,这使得DMTA循环次数减少,可以加速胺基甲酸酯 Cbl-b 抑制剂的发现。证明了即使在AI结构生成作为药物设计关键技术的时代,SPARK依然是可靠的、高效的生物等排体替换技术。
方法与结果
首先,将NX-1607与Cbl-b的共晶结构PDB 8GCY下载到Flare,用Protein Prep进行标准的结构准备。然后将其中的共晶配体NX-1607提取到配体表单,作为SPARK生物等排体替换实验的起点分子(Starter)。
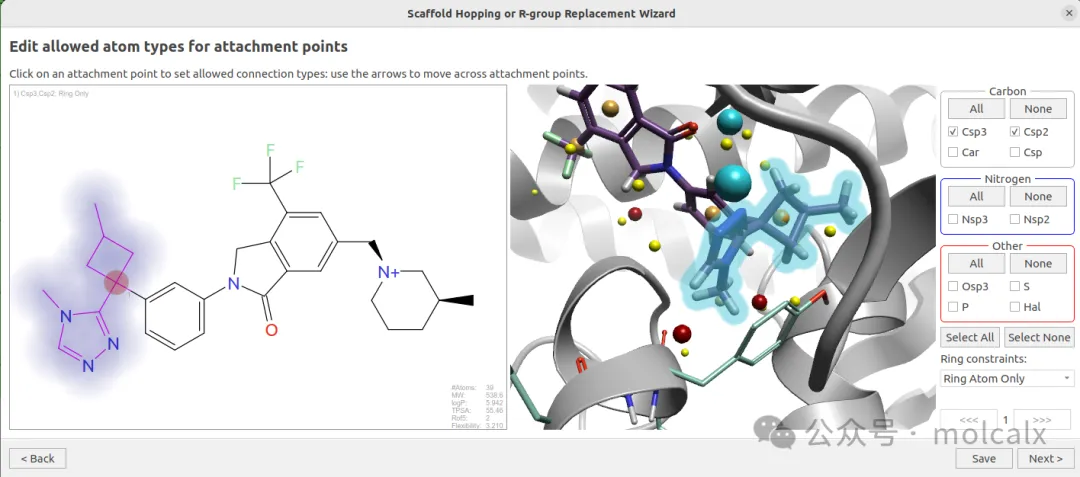
图6. SPARK生物等排体替换的图形界面
然后,在SPARK生物等排体替换向导图形界面中(图6),用鼠标从NX-1607的2D图中选中要被替换的部分(图6高亮显示的部分),并设定连接点原子的类型。这里,我设置为:Csp3, Ring Atom Only.
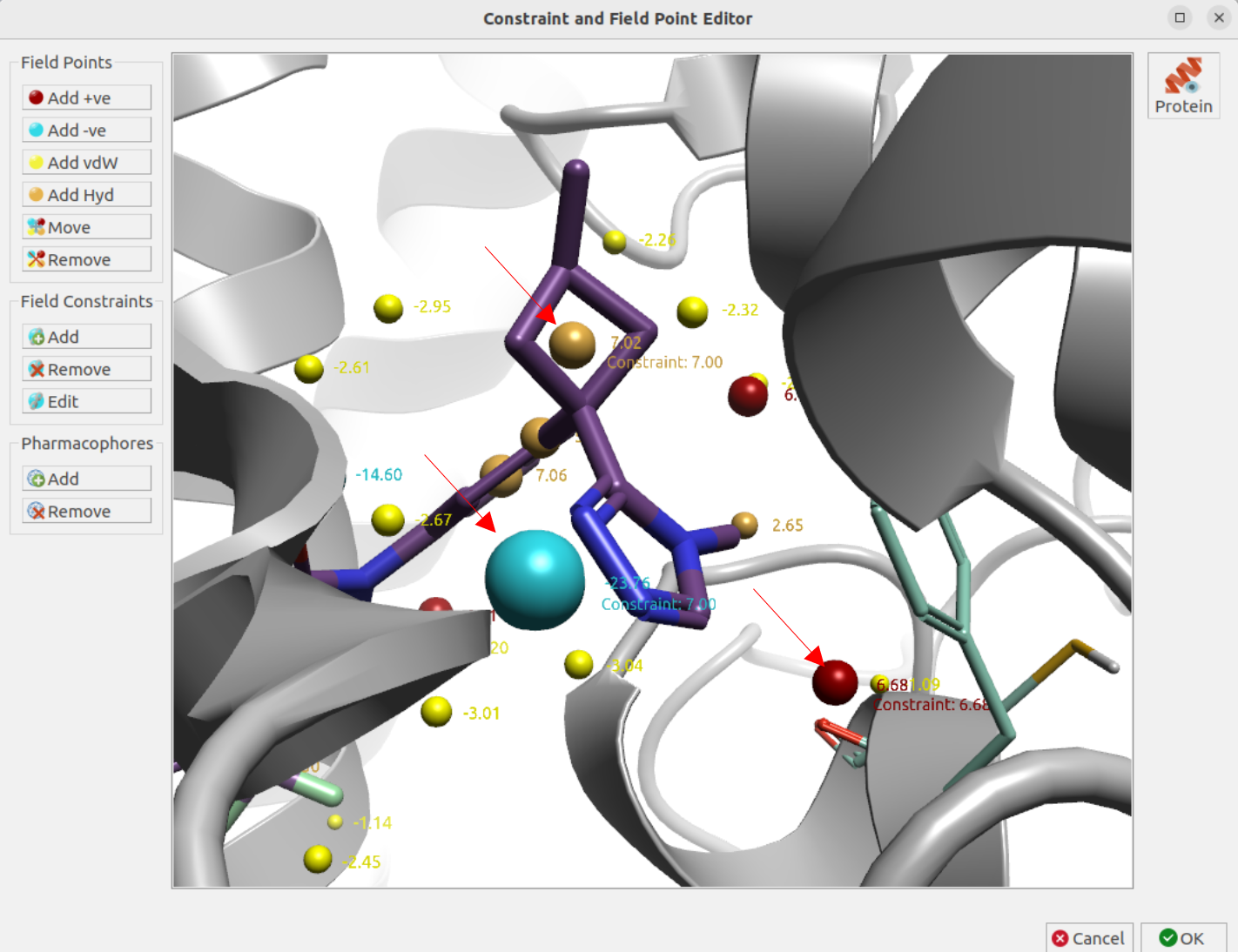
图7. 药效团与场点约束
为了让生物等排体替换的结果更加理想,根据之前的相互作用分析设置了如图7的红色箭头高亮所示的场点约束。第一个是靠近三氮唑两个氮原子的蓝色负场点,表示配体在这里要有强的负静电场或氢键受体基团;第二个约束是金黄色的疏水场点,表示需要配体在这个位置是个疏水基团;第三个是靠近TYR363羰基氧原子的红色正场点,表示需要配体在这个位置要有强的正静电场或者是氢键供体基团;最后一个约束是在1,2,4-三氮唑环上的C-H氢原子位置设置氢键供体,这个约束强调了需要用经典的氢键供体。
图8. 参比分子与蛋白的设置
在进行生物等排体替换时,还设置将共晶的蛋白结构作为排除体积,以约束生成分子的形状不得与共晶蛋白的原子发生空间立体冲突。这里仅将A链作为排除体积,如图8所示。
图9. 数据库的选择
生物等排体从数据中搜索而来。这里,我选择了四个数据库:COD,ChEMBL_common,VeryCommon与Common等四个数据库,共包含177734个化合物片段,如图9所示。
图10. 高级选项
采用基于配体相似性打分函数对片段库进行搜索(图10),仅保留打分最高的500个分子,为了在结果里避开1,2,4-三氮唑结构,还在Advanced Filters设置排除1,2,4-三氮唑子结构与排除含有带正电的氮原子与带负电氧原子,其它参数全部采用默认值。
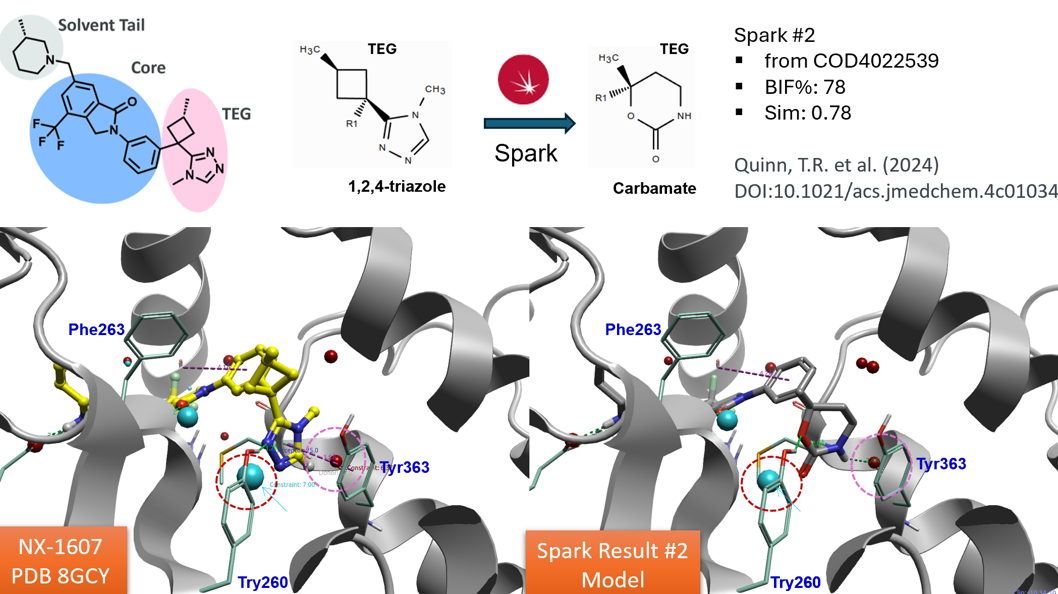
Spark计算生成了500个结果,每个结果的生物等排体指数(BIF%)都大于50,与起点分子的3D相似性打分在0.509-0.972之间。我们注意到,在500个结果中,排名第2的TEG生物等排体就是Quinn等人7发现的化合物12的氨基甲酸酯(carbamate),如图11所示。

图11. Spark#2(左)就是化合物12的氨基甲酸酯(carbamate,右)
Spark#2的氨基甲酸酯片段来源于COD数据库(COD ID:4022539)。以PDB 8GCY的共晶配体NX-1607为参比分子,SPARK#2(Carbamate)的生物等排体指数(BIF%)= 78,综合场点相似性打分Sim=0.781,其中形状场相似性Ssim=0.779以及静电场相似性Fsim=0.782。从图11可以看到,1,2,4-三氮唑两个添加约束的场点很好的被SPAKR#2重现出来。
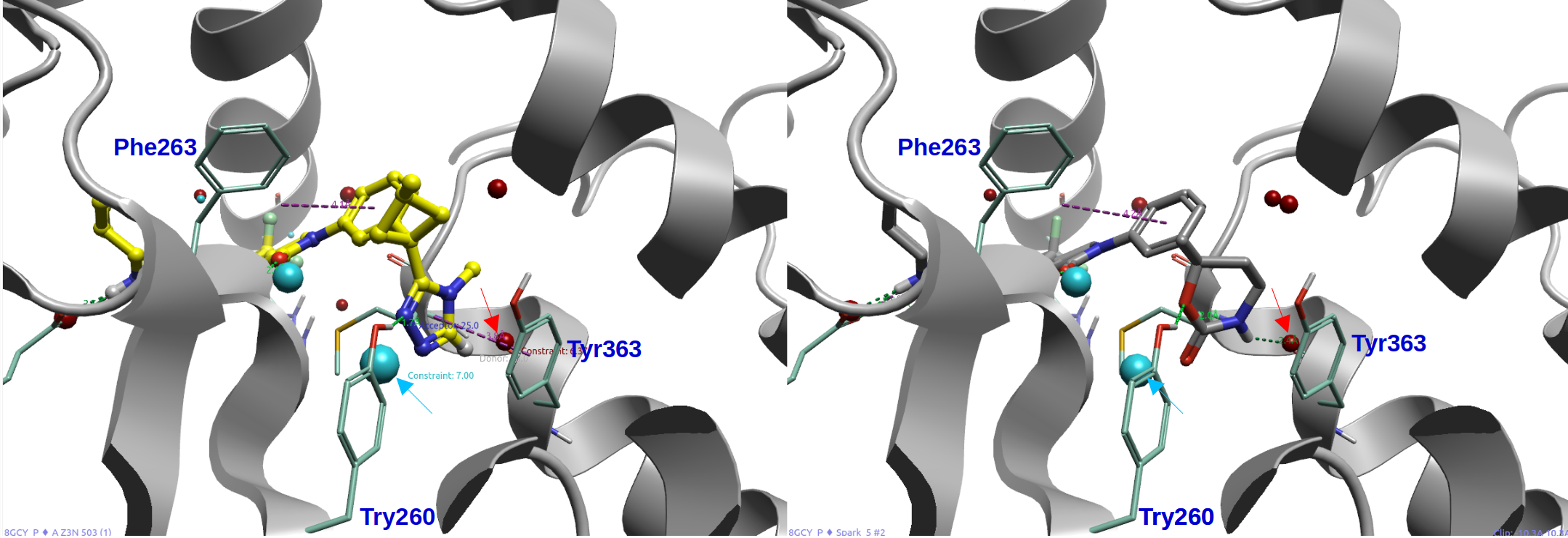
图12. 两种TEG的静电场点与相互作用模式比较。左:1,2,4-三氮唑(左);右:Spark#2的氨基甲酸酯(carbamate,右);灰色飘带:Cbl-B。
图12进一步在CBL-B的结合口袋里比较了1,2,4-三氮唑与Spark#2这两种TEG的场点与结合模式。1,2,4-三氮唑的一个N原子作为氢键受体与Tyr260发生氢键作用,这个相互作用可被靠近该氮原子的一个蓝色-ev场点(蓝色箭头高亮所指)所表征,这与Spark#2的氨基甲酸酯片段同样位置的一个蓝色-ev场点(蓝色箭头高亮所指)相对应。1,2,4-三氮唑的C-H方向上有一个红色的+ev场点(红色箭头高亮所示)与Tyr363的羰基氧相重合,这被Spark#2的氨基甲酸酯的酰胺NH片段重现,与Tyr363碳基氧附近同样位置的一个红色+ev场点(红色箭头高亮所指)相对应。所不同的是,1,2,4-三氮唑与Tyr363发生的π-π相互作用没有被Spark#2的氨基甲酸酯重现,但是后者的经典氢键相互作用可能强于前者的C─H···O=C氢键相互作用而得以补偿。
总的来说,使用Spark的生物等排体替换实验,可以从NX-1607的1,2,4-三氮唑TEG开始一步直接设计出Spark#2的氨基甲酸酯,跳过了Quinn等人7第1轮与第2轮DMTA循环而直接进入第3轮DMTA循环,可以加速胺基甲酸酯 Cbl-b 抑制剂的发现。
结论
Cbl-b抑制剂与Tyr363相互作用的TEG片段长期以来都保留了1,2,4-三氮唑结构,直到阿斯利康Quinn等人7采用生成式AI与SBDD相结合,经过多轮的DMTA循环最后发现新型TEG——六员环的胺基甲酸酯(Carbamate)。在本文中,我们采用Spark基于配体相似性打分对1,2,4-三氮唑进行生物等排体替换,一步计算就从结果里以排名第2的方式找到了六员环的胺基甲酸酯,重现了阿斯利康Quinn等人7化合物12的TEG设计。这使得DMTA循环次数减少,可以加速胺基甲酸酯 Cbl-b 抑制剂的发现。这说明了,即使生成式AI与人类专家都难以实现的跃迁,Spark依然可以提供操作简单、结果可靠、高效的生物等排体替换方案。
文献
- Swaminathan, G. and Tsygankov, A.Y. (2006) “The Cbl family proteins: Ring leaders in regulation of cell signaling,” Journal of Cellular Physiology. Available at: https://doi.org/10.1002/jcp.20694.
- Augustin, R.C., Bao, R. and Luke, J.J. (2023) “Targeting Cbl-b in cancer immunotherapy,” Journal for ImmunoTherapy of Cancer. Available at: https://doi.org/10.1136/jitc-2022-006007.
- Bachmaier, K. et al. (2000) “Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b,” Nature, 403(6766). Available at: https://doi.org/10.1038/35003228.
- Barsanti, P. A. et al. (2019) “Inhibitors of CBL-B and methods of use thereof,” WO 2019148005 A1
- Discovery and optimization of Cbl-b inhibitors. https://www.nurixtx.com/wp-content/uploads/2022/09/Nurix-CBL-B-DOT-Talk_JK.pdf (accessed Sept 06, 2023).
- Cohen, F. et al. NX-1607: a First-In-Class Inhibitor of Casistas B-lineage Lymphoma B (CBL-B) for Immuno-Oncology. Abstracts, ACS Spring Meeting 2024, New Orleans, LA, March 20, 2024.
- Quinn, T.R. et al. (2024) “Accelerated Discovery of Carbamate Cbl-b Inhibitors Using Generative AI Models and Structure-Based Drug Design,” Journal of Medicinal Chemistry, 67(16), pp. 14210–14233. Available at: https://doi.org/10.1021/acs.jmedchem.4c01034.
- Spark Version 10.7.http://www.cresset-group.com/software/spark
联系我们
想在自己的项目中亲自使用SPARK进行生物等排体替换,请联系我们获取免费的试用版;或者联系我进行在线演示;你还可以采购软件或委托研究与我们进行项目合作。
- 电邮:info@molcalx.com
- 电话:020-38261356
原创文章,作者:小墨,如若转载,请注明出处:《用SPARK生物等排体替换加速胺基甲酸酯 Cbl-b 抑制剂的发现》http://blog.molcalx.com.cn/2025/01/22/cbl-b-inhibitor-teg-scaffold-hopping.html


