
摘要:本文探讨结构生物学在药物发现中的应用及挑战。通过X-衍射晶体学、冷冻电镜等技术获取生物分子与小分子复合物结构,为药物设计提供关键指导。结构测定方法如浸泡法便捷但可能失真,共结晶法虽耗时却能更准确反映结合构象。然而,结构解析受限于实验条件(低温、非生理溶液)、分辨率及电子密度图建模的主观性,需结合分子动力学模拟揭示动态相互作用。验证结构需通过突变实验(如EGFR-T790M耐药突变研究)或配体修饰,证明关键氢键或疏水作用对活性的影响。研究强调非极性范德华相互作用常被低估,需结合多维度可视化与计算工具全面评估。结构生物学数据虽能加速先导化合物优化,但其解读必须结合生化实验验证,并在现有结构数据库背景下审慎分析,避免过度依赖单一静态结构。该领域需平衡结构指导的创新与实验验证的严谨性,以充分发挥其在药物研发中的核心价值。
原文:Heppner, D.E. (2025) “Ascertaining a Structural Basis in Drug Discovery and Development,” Journal of Medicinal Chemistry, 68(5), pp. 4991–4995. Available at: https://doi.org/10.1021/acs.jmedchem.5c00326.
编译:肖高铿
药物化学家常常利用“结构生物学基础(structural basis)”来理性理解小分子的活性,并在药物发现中用来指导基于结构的优化1-3。具备结构生物学信息支持的项目,也就是拥有用实验方法确定的候选药物结合于目标靶点三维结构的研究项目,能够以更快的速度推进,从而加速对苗头化合物或先导化合物的优化。然而,通过视觉上吸引人的平台解读这些信息并不总是那么简单,深思熟虑的解释还需要进一步的实验以及比较性结构分析,从而为下一步可操作的决策提供依据。
“结构生物学基础”这一术语涵盖了生物化学中的各种场景。例如,理解酶调控是如何在影响生物分子结构变化的改变或相互作用的背景下发生的。在本文中,重点在于理解小分子如何与生物分子受体靶点结合。在我自己的研究中4-10,通过X-衍射晶体学获取结构通常成为新问题的起点——这些问题只能通过精心设计的结合干扰或通过已报道的结构数据与其他化合物进行比较来解答。考虑到J. Med. Chem.和ACS Med Chem Lett.关于“药物发现与开发中的结构生物学”虚拟专刊的重点,反思结构生物学信息的用途及其在解释过程中遇到的各种复杂问题是非常合适的11,12。通过探索这些数据如何被解读和扩展应用,我们可以增强其在药物发现中的作用,并对未来的科研产生有意义的影响。
获取结构
从头开始追求生物分子的三维结构是一项艰巨的任务。这一过程涉及生产并纯化出稳定且均一的生物分子(说起来容易做起来难),随后需要寻找适合进行结构解析的实验条件。关于此类协议(protocols)和方法的策略,已有大量文献报道,涵盖了最常使用的技术:核磁共振(NMR)、X-衍射晶体学以及冷冻电子显微镜(cryo-EM)等等13-17。
人们普遍认为,每个结构生物学项目通常都是通过独特且带有个性化特点的方法来实现的。事实上,当某个备受关注的生物分子的首个结构被报道时,除了结构生物学本身提供的见解之外,最引人入胜的方面之一是研究人员用于实现这一目标的方法学。在我看来,我们领域的所有成员都必须认识到,那些以结构生物学为基础的项目是建立在前人往往艰辛的突破之上的,正是他们的创新方法使得理解药理学上重要的小分子的结构基础成为可能。
当然,这个故事还有另一半,因为小分子化合物(也称为配体)也需要被纳入考量。药物化学家通常解读的结构来源于X-衍射晶体学,这些结构是通过两种方法获得的:一种是将化合物暴露于预先制备的晶体中,这种方法被称为“浸泡(soaking)”;另一种是将化合物与生物分子预孵育后,再将混合物置于结晶条件下,这种方法被称为“共结晶(cocrystallization)”18-19。浸泡法通常更简单,并且适合对多种不同化合物进行筛选,因为这种方法通常从已经具备衍射质量的晶体开始。而共结晶则可能需要筛选独特的结晶条件——这在时间和资源上都是昂贵的工作——并且不一定能保证成功。“共晶(cocrystal)”一词指的是在单一晶格中包含两种或多种分子化合物。共晶结构是指任何观察到的配体与生物分子结合的结果,例如酶底物或抑制剂化合物的结合,而不一定是通过共结晶方法获得的结构。由于化合物的结合可能会导致靶标结构的变化,而这种变化在用于浸泡的晶体中并不常见,因为生物分子在晶体中“堆积”的方式不同,因此来自共结晶的结构通常更能准确反映化合物结合模式中最有利的构象。
这些差异的重要性可以通过比较药物AZD9291(奥希替尼)与表皮生长因子受体(EGFR)激酶域结合时,通过浸泡或共结晶获得的X-衍射共晶结构来说明。这两种方法导致了药物完全不同的构象,而通过共结晶获得的结构反映了最相关的结合模式20。在药物发现的背景下,令人沮丧的是,在结构生物学中此类差异性发现是很常见的,这也强烈促使研究人员进行实验以验证结构测定中观察到的结果。
有了结构,接下来作什么?
观察到一个新的分子与蛋白质靶标结合,是科学研究中最令人欣慰的里程碑之一——这是我个人经验之谈4,5,7。 一旦得到化合物的模型且结构经过适当优化,这往往代表了揭示长期假设答案的“真相时刻(moment of truth)”,包括结构-活性关系的研究成果、预期策略是否实现了预期的相互作用,以及关于如何最有效地优化该化合物的初步见解。在所有情况下,关键信息包括靶标与配体之间的相互作用、化合物的结合几何以及其他细节。此外,化合物可能仅与靶标受体的特定构象结合,从而提供进一步的见解。尽管结构数据具有强大的作用,但仍需验证某些相互作用对化合物活性谱的重要性,以正确推动项目的进展21。
在这方面,生化结合或活性测定是不可或缺的,它们是确定结构基础的必要手段。这些实验的目标应该是实现对变量的精确、头对头比较,从而突出影响化合物结合的特定相互作用的重要性。在结合模式上的差异可以通过多种方法进行实验验证,这些方法可以测量可逆结合的参数(例如,IC50、Ki、Kd、ΔG)以及共价结合配体的参数(例如,不可逆结合的kinact/KI,可逆共价结合的Ki*),使用纯化的生物分子进行测定。近年来,在细胞环境中进行的诸如NanoBRET22(或类似方法23)之类的靶标占据测试变得越来越重要。
为了验证相互作用的重要性,可以通过干扰生物分子靶标和/或化合物本身的某些方面来进一步表征结合差异。对于蛋白质靶标,引入突变(例如改变侧链基团)可以帮助评估体系中某些结构成分的影响,特别是当突变导致结合减弱或丧失时。一个著名的例子来自于针对非小细胞肺癌靶向治疗相关的EGFR酪氨酸激酶激活突变的药物研究24。一种二次突变,即T790M “gatekeeper” 突变,会导致对第一代和第二代酪氨酸激酶抑制剂(TKIs)治疗的耐药性25。当前FDA批准的药物奥希替尼(osimertinib)和拉泽替尼(lazertinib)通过与T790M侧链直接且有利的结合,有效抑制了这种突变型EGFR激酶,这种结合是通过X-衍射共晶结构中观察到的疏水范德华相互作用实现的(如图1A中的拉泽替尼)6,20。这种相互作用的相关性是通过比较含T790M突变和野生型(WT)EGFR激酶的时间依赖性动力学不可逆抑制速率常数而得以确认6,26,27。
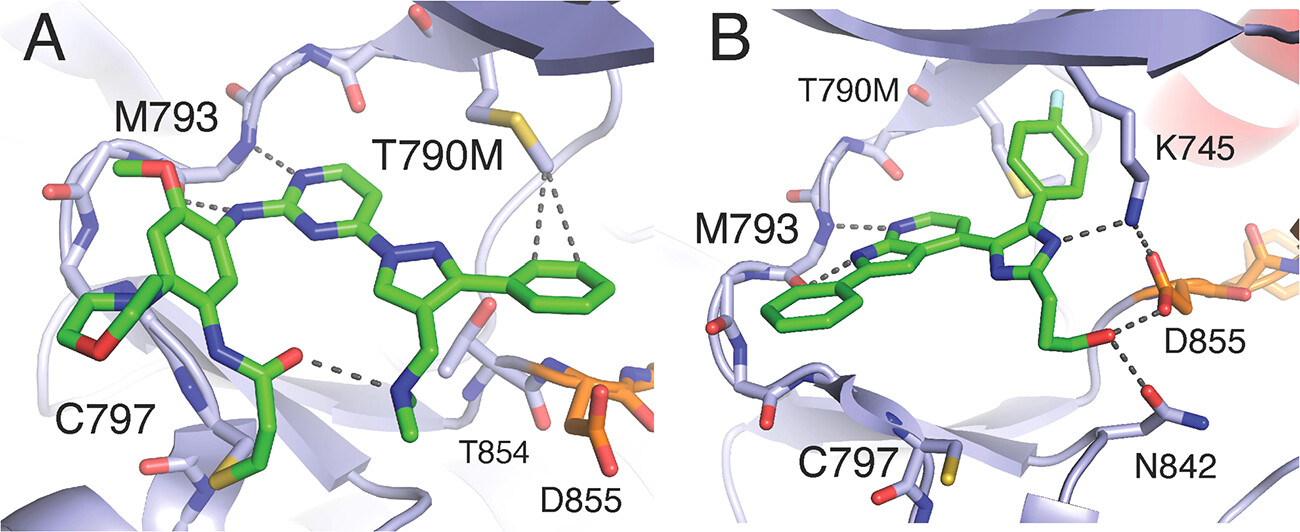
图1. 小分子酪氨酸激酶抑制剂对EGFR激酶域实现特异性靶向的分子间相互作用。A)拉泽替尼(Lazertinib)与含T790M突变的EGFR复合物显示出范德华相互作用,这与其对T790M依赖性耐药的选择性一致(PDB ID 7UKW)。改编自参考文献(6),经许可使用。版权归属于2022年美国化学学会。B)三取代咪唑LN2084与EGFR的复合物,显示了与保守催化赖氨酸K745的氢键作用(PDB ID 6V5N)。改编自参考文献(5),经许可使用。版权归属于2020年美国化学学会。
另一方面,有些相互作用无法通过干扰生物分子来验证。例如,锚定在主链酰胺或对酶活性至关重要的保守残基上的化合物会带来挑战,因为对这些蛋白质区域的任何改变都会破坏其典型的结构和功能。我们研究过的例子包括EGFR三取代咪唑类TKIs的X-衍射共晶结构,显示了配体与催化赖氨酸残基(K745,图1B)之间的氢键5,8,28。 通过突变方法验证这种相互作用是不现实的,因为该位点的突变会破坏酶的功能。取而代之的是,我们制备了在咪唑氮原子上具有特定N-甲基化的衍生物,其中一个设计专门用于阻断K745的氢键作用。这使得我们可以评估X-衍射共晶结构中观察到的赖氨酸氢键作用,从而揭示了第四代EGFR TKIs如何通过更强的激酶域结合克服C797S突变的结构基础5,28。尽管此方法在本案例中有效,但重要的是不能忽视改变化合物结构可能带来的潜在不良后果,特别是在母核杂环上添加额外的空间位阻可能导致意外的排斥相互作用。
结构测定的“现实”:眼见真的为实吗?
药物化学家需要重视的另一个关键点是,结构坐标并非绝对明确无误。结构测定方法对分子要求很高,并且通常在与生理环境显著不同的条件下进行。例如,蛋白质通常在含有高浓度的多种沉淀剂的特殊溶液中结晶,并且pH值被缓冲到远离生理范围。此外,大多数X-衍射晶体学和冷冻电子显微镜(Cryo-EM)结构是在低温(约100 K)下获得的。这种低温可能会影响化合物的结合相互作用以及蛋白质的构象,与酶活性测定或药理条件相关的较高温度下的情况不同。对此,室温X-衍射晶体学等方法试图评估温度的影响29;然而,由于这些技术在实验上比低温方法更具挑战性,其应用仍然相对有限。当然,结构的分辨率(即高分辨率极限)可能导致原子坐标存在潜在的不确定性,包括氨基酸侧链和溶剂水分子的位置。需要特别注意的是,在项目中应密切关注相关结构的分辨率,因为它们可能会有很大差异。
在我们自己的研究中,我们观察到,在将X-衍射共晶结构与微秒时间尺度的分子动力学(MD)模拟进行比较时,蛋白质-配体相互作用存在显著差异(图2)9。通过X-衍射共晶结构,化合物1和2与EGFR K745残基的相互作用表明,该侧链会结合到配体内的不同基团,即从图2A中的ATP侧摆动到图2C中的别构位点,这一现象单独来看似乎对解释这些化合物之间的巨大结合差异至关重要。然而,MD模拟提供了不同的视角,表明在X-衍射共晶结构中观察到的K745氢键实际上是由水分子介导的。这种明显脱节的原因尚不清楚,且超出了原研究的范围。根据前一段提到的观点,可以很容易推测,X-衍射结构的低温条件导致了与较高温度下观察到的不同相互作用,而MD模拟正是设计用来模拟较高温度下的情况,或者是因为这些结构的分辨率不够高,无法观察到介导这些氢键的水分子。
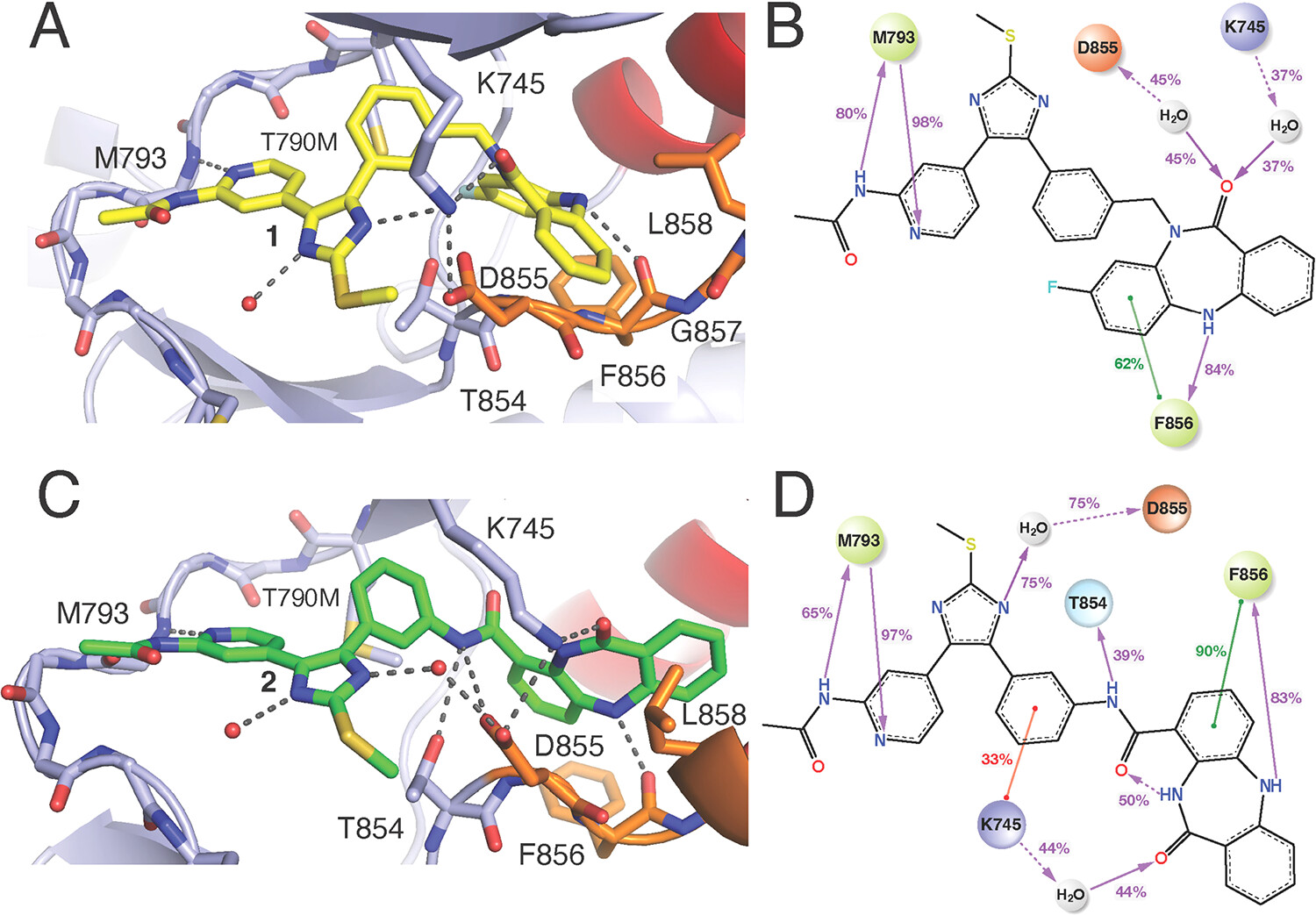
图2. EGFR激酶蛋白与双价激酶抑制剂之间可供选择的替代相互作用。分别基于化合物1(A和B,PDB ID 8FV3)和化合物2(C和D,PDB ID 8FV4)的X-衍射共晶结构以及基于分子动力学(MD)模拟得到的相互作用频率。B和D:仅显示在模拟时间中出现超过20%的相互作用,详细信息可参见文献(9)。注意配体与铰链区M793之间的类似氢键相互作用,而在共晶结构中观察到K745与配体形成直接氢键,在MD模拟中这些氢键被发现是由水分子介导的。转载自参考文献(9),遵循CC-BY 4.0许可协议。版权归属于2024年Wittlinger, F.; Ogboo, B. C. 等人。
另一个关于理解结构的关键点是,需要认识到X-衍射晶体学和冷冻电镜结构的坐标是从电子密度图的主观建模中推导出来的30。这一点对于药物化学领域的资深研究者和学生来说都是重要的一课,因为在可视化分析时从蛋白质数据库(PDB)中获取的结构通常并未附带用于推导坐标的密度图。常见的风险之一是过度解读某些原子位置,特别是对于那些柔性且与靶受体锚定较弱的官能团。Lamb、Kappock和Silvaggi撰写的一篇优秀综述描述了各种场景及相关数值术语(例如原子B因子)31,这为正确分析密度图提供了指导,这些建议最适合在研究者有机会进行配体定位、模型构建和优化时的参考30。使用免费的可视化软件Coot,通过“使用EDS获取PDB和密度图(fetch PDB and Map using EDS)”功能,可以轻松理解和访问坐标及密度图32-33。借助这些工具,药物化学家和药物研发研究人员可以对PDB中的结合模式更有信心,并利用这些发现进行基于结构的设计以及理解化合物活性的结构基础。
保持全局的视角
如上所述,大多数研究者,尤其是学生,最初是通过图形程序(如Chimera或PyMOL)直接从蛋白质数据库(PDB)中可视化化合物-受体结构的坐标来接触结构的34。我们大多数人习惯于关注更具离子型相互作用,包括氢键,因为这些可以通过测量原子间距离来可靠评估21。然而,在药物化学课程中教授结构解析的基本原理,或在我们研究小组的背景下,我越来越强调那些不太直观的非离子型范德华相互作用的重要性和价值。
这显然是一个需要根据具体情况分析的问题,某些非极性相互作用在结构中尤为突出(例如图1A),但是这些较弱的非极性相互作用常常被忽视,尽管大多数化合物由疏水基团组成。我们研究过的一个例子9源于对EGFR中ATP别构双价抑制剂(AABIs)产生显著活性差异的相互作用和物理性质的理解(图2)。最初,氢键的差异显著且看似易于解释,表明这些相互作用的增强推动了观察到的结合改善(PDB ID:8FV3和8FV4)。然而,随后的分子动力学(MD)模拟揭示,结合的增强实际上是更强的极性和非极性范德华相互作用共同作用的结果。回顾来看,由于氢键和水介导相互作用的变化非常明显,较强的非极性相互作用的作用被我们忽略了。一种有助于更好地理解非极性相互作用的方法是使用多种可视化风格生成结合模式。通过在“棍棒”、“球体”和“表面”等原子显示风格之间切换,可以更简单地评估这些较弱的相互作用。例如,“棍棒”表示可能会人为地让观察者感觉原子之间存在显著的空间,而实际上当以“球体”形式查看时往往并非如此。这强调了整合计算工具(如MD模拟)的重要性,以补充结构可视化,并提供对这些微妙但关键的相互作用更全面的理解。
总结思考
尽管本文仅触及了结构生物学数据用途的表面,但显而易见的是,生成一个有用的结构基础对于药物发现至关重要,但也并非没有不确定性。结构生物学结果的可信度受到多种混杂因素的影响,例如化合物与靶标受体接触的方法(如浸泡与共结晶)、远离生理条件的实验环境(温度、溶液条件)、数据质量(分辨率限制)以及电子密度图的主观建模。由这些因素引发的问题无疑会影响药物发现,并对计算机虚拟对接工作产生深远影响35。因此,进行支持性实验以确认这些结构细节的重要性至关重要,包括利用干扰靶标(如突变实验)或配体结构改变进行的生化结合测量。重要的是,应在PDB和文献中已有结构的背景下生成化合物的结构基础,以便更好地评估新化合物结构相对于已公开结构的优势和劣势。话虽如此,也应警惕对某些结构基础相互作用的重要性采取过于严格的观点,因为这可能会限制对那些可能意外带来更有前景特性的药物结构的探索。结构生物学信息在药物发现中的价值毋庸置疑,结构基础的开发必须通过对实验变量的深思熟虑分析以及对维持受体-配体复合物相互作用的功能验证来进行指导。
文献
- Wei, H.; McCammon, J. A. Structure and dynamics in drug discovery. npj Drug Discovery 2024, 1 (1), 1.
- Arrowsmith, C. H. Structure-guided drug discovery: back to the future. Nature Structural & Molecular Biology 2024, 31 (3), 395−396.
- Congreve, M.; Murray, C. W.; Blundell, T. L. Keynote review: Structural biology and drug discovery. Drug Discovery Today 2005, 10 (13), 895−907.
- De Clercq, D. J. H.; Heppner, D. E.; To, C.; Jang, J.; Park, E.; Yun, C.-H.; Mushajiang, M.; Shin, B. H.; Gero, T. W.; Scott, D. A.; et al. Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS Med. Chem. Lett. 2019, 10 (11), 1549−1553.
- Heppner, D. E.; Günther, M.; Wittlinger, F.; Laufer, S. A.; Eck, M. J. Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem. 2020, 63 (8), 4293−4305.
- Heppner, D. E.; Wittlinger, F.; Beyett, T. S.; Shaurova, T.; Urul, D. A.; Buckley, B.; Pham, C. D.; Schaeffner, I. K.; Yang, B.; Ogboo, B. C.; et al. Structural Basis for Inhibition of Mutant EGFR with Lazertinib (YH25448). ACS Med. Chem. Lett. 2022, 13 (12), 1856−1863.
- Wittlinger, F.; Heppner, D. E.; To, C.; Günther, M.; Shin, B. H.; Rana, J. K.; Schmoker, A. M.; Beyett, T. S.; Berger, L. M.; Berger, B.-T.; et al. Design of a “Two-in-One” Mutant-Selective Epidermal Growth Factor Receptor Inhibitor That Spans the Orthosteric and Allosteric Sites. J. Med. Chem. 2022, 65 (2), 1370−1383.
- Bhattacharjee, D.; Bakar, J.; Chitnis, S. P.; Sausville, E. L.; Ashtekar, K. D.; Mendelson, B. E.; Long, K.; Smith, J. C.; Heppner, D. E.; Sheltzer, J. M. Inhibition of a lower potency target drives the anticancer activity of a clinical p38 inhibitor. Cell Chemical Biology 2023, 30 (10), 1211−1222 e1215, accessed 2024/07/11.
- Wittlinger, F.; Ogboo, B. C.; Shevchenko, E.; Damghani, T.; Pham, C. D.; Schaeffner, I. K.; Oligny, B. T.; Chitnis, S. P.; Beyett, T. S.; Rasch, A.; et al. Linking ATP and allosteric sites to achieve superadditive binding with bivalent EGFR kinase inhibitors. Communications Chemistry 2024, 7 (1), 38.
- Wittlinger, F.; Chitnis, S. P.; Pham, C. D.; Damghani, T.; Patel, K. B.; Mollers, M.; Schaeffner, I. K.; Abidakun, O. A.; Deng, M. Q.; Ogboo, B. C.; Rasch, A.; Beyett, T. S.; Buckley, B.; Feru, F.; Shaurova, T.; Knappe, C.; Eck, M. J.; Hershberger, P. A.; Scott, D. A.; Brandt, A. L.; Laufer, S. A.; Heppner, D. E. Tilting the Scales toward EGFR Mutant Selectivity: Expanding the Scope of Bivalent “Type V” Kinase Inhibitors. J. Med. Chem. 2024, 67 (23), 21438−21469.
- Heppner, D. E.; Bigi-Botterill, S. V.; Riley, A. P.; Chibale, K.; Lindsley, C. W. Structural Biology in Drug Discovery and Development: Call for Papers. ACS Med. Chem. Lett. 2024, 15 (8), 1167−1168.
- Heppner, D. E.; Bigi-Botterill, S. V.; Riley, A. P.; Chibale, K.; Lindsley, C. W. Structural Biology in Drug Discovery and Development: Call for Papers. J. Med. Chem. 2024, 67 (15), 12461−12462.
- Hu, Y.; Cheng, K.; He, L.; Zhang, X.; Jiang, B.; Jiang, L.; Li, C.; Wang, G.; Yang, Y.; Liu, M. NMR-Based Methods for Protein Analysis. Anal. Chem. 2021, 93 (4), 1866−1879.
- Budziszewski, G. R.; Snell, M. E.; Wright, T. R.; Lynch, M. L.; Bowman, S. E. J. High-Throughput Screening to Obtain Crystal Hits for Protein Crystallography. JoVE 2023, No. 193, No. e65211.
- Kermani, A. A. A guide to membrane protein X-ray crystallography. FEBS Journal 2021, 288 (20), 5788−5804.
- McPherson, A.; Gavira, J. A. Introduction to protein crystallization. Acta Crystallographica Section F 2014, 70 (1), 2−20.
- Weissenberger, G.; Henderikx, R. J. M.; Peters, P. J. Understanding the invisible hands of sample preparation for cryo-EM. Nat. Methods 2021, 18 (5), 463−471.
- Wienen-Schmidt, B.; Oebbeke, M.; Ngo, K.; Heine, A.; Klebe, G. Two methods, one goal: Structural differences between cocrystallization and crystal soaking to discover ligand binding poses. ChemMedChem. 2021, 16 (1), 292−300.
- Kašcá̌ ková, B.; Koutská, A.; Burdová, M.; Havlíčková, P.; Kutá Smatanová, I. Revealing protein structures: crystallization of protein-ligand complexes – co-crystallization and crystal soaking FEBS Open Bio 2024, DOI: 10.1002/2211-5463.13913.
- Yan, X.-E.; Ayaz, P.; Zhu, S.-J.; Zhao, P.; Liang, L.; Zhang, C. H.; Wu, Y.-C.; Li, J.-L.; Choi, H. G.; Huang, X.; et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63(15), 8502−8511.
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53 (14), 5061−5084.
- Machleidt, T.; Woodroofe, C. C.; Schwinn, M. K.; Méndez, J.; Robers, M. B.; Zimmerman, K.; Otto, P.; Daniels, D. L.; Kirkland, T. A.; Wood, K. V. NanoBRET� A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10 (8), 1797−1804.
- Shirley, J. D.; Gillingham, J. R.; Nauta, K. M.; Diwakar, S.; Carlson, E. E. kinact/KI Value Determination for Penicillin-Binding Proteins in Live Cells. ACS Infectious Diseases 2024, 10 (12), 4137−4145.
- Heppner, D. E.; Eck, M. J. A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Sci. 2021, 30(8), 1535−1553.
- Yun, C.-H.; Mengwasser, K. E.; Toms, A. V.; Woo, M. S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (6), 2070−2075 (acccessed 2024/10/03).
- Patel, K. B.; Heppner, D. E. Lazertinib: Breaking the mold of third-generation EGFR inhibitors. RSC Medicinal Chemistry 2025, DOI: 10.1039/D4MD00800F.
- Hoyt, K. W.; Urul, D. A.; Ogboo, B. C.; Wittlinger, F.; Laufer, S. A.; Schaefer, E. M.; May, E. W.; Heppner, D. E. Pitfalls and Considerations in Determining the Potency and Mutant Selectivity of Covalent Epidermal Growth Factor Receptor Inhibitors. J. Med. Chem. 2024, 67 (1), 2−16.
- Damghani, T.; Wittlinger, F.; Beyett, T. S.; Eck, M. J.; Laufer, S. A.; Heppner, D. E. Chapter Six – Structural elements that enable specificity for mutant EGFR kinase domains with next-generation small-molecule inhibitors. In Methods in Enzymology; Richard, J. P., Moran, G. R., Eds.; Vol. 685; Academic Press, 2023; pp 171−198.
- Fraser, J. S.; van den Bedem, H.; Samelson, A. J.; Lang, P. T.; Holton, J. M.; Echols, N.; Alber, T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (39), 16247−16252.
- Lamb, A. L.; Kappock, T. J.; Silvaggi, N. R. You are lost without a map: Navigating the sea of protein structures. Biochimica et Biophysica Acta (BBA) – Proteins and Proteomics 2015, 1854 (4), 258−268.
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M. T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119 (3), 1626−1665.
- Emsley, P.; Lohkamp, B.; Scott, W. G.; Cowtan, K. Features and development of Coot. Acta Crystallographica Section D 2010, 66(4), 486−501.
- Kleywegt, G. J.; Harris, M. R.; Zou, J.-y.; Taylor, T. C.; Wahlby, A.; Jones, T. A. The Uppsala Electron-Density Server. Acta Crystallographica Section D 2004, 60 (12 Part 1), 2240−2249.
- Burley, S. K. Impact of structural biologists and the Protein Data Bank on small-molecule drug discovery and development. J. Biol. Chem. 2021, 296, 100559.
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M. A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64 (5), 2489−2500.


