
摘要:本文介绍了结合模式元动力学(Binding Pose MetaDynamics,BPMD)模拟的开源python实现OpenBPMD,演示了Flare与OpenBPMD联合应用可大幅降低计算门槛,为研究者提供了一个能快速构建研究体系并高效完成蛋白-配体结合模式稳定性评估的完整工作流。具体而言,本文以微管蛋白α-1β与Epothilone A(EpoA)结合模式为例,展示了Flare与OpenBPMD联合使用在评估蛋白质-配体结合模式稳定性中的强大能力。结果表明,Flare以非常友好、易用的方式提供了MetaD模拟所需的输入文件;而OpenBPMD能够有效区分实验争议结构中稳定与不稳定的结合模式:对于早期因实验方法受限而被质疑的PDB 1TVK模型,其CompScore(26.964)和PoseScore(27.697 Å)显著偏离稳定阈值(PoseScore \(<\) 2.0 Å),且接触持续性(ContactScore = 0.147)极低;而分辨率高的结构PDB 4I50则表现出优异的稳定性(PoseScore = 1.401 Å,ContactScore = 0.710),充分验证了OpenBPMD打分与实验观测的一致性。这一发现不仅验证了Flare与OpenBPMD联用的可靠性,也为解决实验结构争议提供了计算生物学视角的佐证。近期OpenBPMD在Protac分子设计中的成功应用,彰显了该方法在复杂分子体系中的拓展潜力。
肖高铿/2025-05-05
前言
具有生物学意义的蛋白质-配体复合物三维结构在基于结构的药物设计(Structure-Based Drug Design, SBDD)中起着关键作用,无论是在苗头化合物发现(hit discovery)阶段,还是在后续的先导化合物优化(lead optimization)阶段中均不可或缺。例如,当前广泛应用的炼金术自由能微扰(Free Energy Perturbation, FEP)方法在预测结合自由能时,也依赖于准确的蛋白质-配体结合模式作为初始输入构象1。因此,评估通过实验手段(如X射线单晶衍射、冷冻电镜等结构生物学技术)或计算方法(如分子对接、AlphaFold3等深度学习预测模型)获得的蛋白质-配体结合模式的正确性,具有重要意义。
在各类计算方法中,分子对接是预测配体结合模式的主要手段之一。然而,目前主流分子对接方法的实际表现仍存在一定局限性。以Wang等人2对10种分子对接程序的系统评估为例,研究结果显示,典型的对接算法仅能在约40%至60%的测试算例中将天然结合模式(定义为与真实结合模式的均方根偏差 RMSD \(<\) 2 Å)排在打分首位;尽管如此,在约60%至80%的测试算例中,在预测的多个结合模式中至少存在一个结合模式可被认为接近天然结合模式。这一结果表明,开发具备更强结合模式排序能力的计算方法对于从分子对接生成的多个结合模式中准确识别真实结合模式不仅有可能而且至关重要。
由Clark等人3于2016年提出的结合模式元动力学(Binding Pose Metadynamics,BPMD)是一种兼具计算速度与精确的结合模式排序方法。BPMD在较短的模拟中对多个候选结合模式进行干扰,然后根据元动力学(Metadynamics,MetaD)模拟过程中观察到的均方根偏差(RMSD,相对于初始配体坐标)以及氢键持续性来评估结合模式稳定性,并对这些结合模式进行排序。最近,Fusani等人4的研究表明,BPMD可以用于识别PDB数据库中正确的结合模式。Miller等人5报道了将BPMD与诸如水分子分析、更长时间的无偏MD模拟以及相对结合自由能计算等其他方法结合以帮助识别天然结合模式。
本文主要目的之一是介绍一款由Lukauskis等人6开发的开源BPMD实现——OpenBPMD。OpenBPMD主要设计用于与分子对接结合使用,以根据构象稳定性对候选结合模式进行重新排序。在该方法中,配体原子会受到元动力学偏置的作用,并依据其稳定性对配体结合模式进行打分。OpenBPMD是一个Python脚本,它利用OpenMM分子动力学引擎7-9来运行元动力学模拟,并结合MDAnalysis10,11和MDTraj12对模拟结果进行处理与分析。为了验证该工具的有效性,Lukauskis等人6将OpenBPMD应用于Clark等人3所使用的数据集上,并得到了与BPMD非常相似的结果。结果发现,在88%的算例中,OpenBPMD能够识别出天然结合模式(\(RMSD < 2 Å\)),并且发现采用基于GCMC/MD的水分子采样方法对溶剂分子进行平衡将有助于获得良好的结果。最近,Ma等人13成功地将OpenBPMD用于Protac项目,用于评估多种不同连接臂Protac的结合模式稳定性,这进一步拓展了OpenBPMD在复杂分子体系中的应用。OpenBPMD的代码是开源的,可免费从GitHub上获取:https://github.com/GervasioLab/OpenBPMD。
本文将以Fusani等人4报道的EpoA-Tubulin共晶结构结合模式的为例,演示如何将强大的药物设计平台Flare14与OpenBPMD相结合,实现快速、便利的配体结合模式稳定性评估。
方法
蛋白结构准备
将待研究的蛋白-配体共晶结构(PDB 1TVK与4I50)从PDB数据库下载到Flare V1014中,并使用其中的Protein Prep工具小心地准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态,任何截短的蛋白质链被封端作为蛋白质准备的一部分。确认结构无误后,将配体从A从蛋白表单中提取到配体表单或将复合物结构(1tvk_prep.pdb,4i50_prep.pdb)导出备用。
AMBER格式MD体系的准备
用Flare/Dynamics来准备OpenBPMD所需的模拟体系的AMBER格式原子坐标(rst7)与拓扑文件(prm7)。不仅可以通过Flare GUI,还可以通过pyflare脚本dynamics.py来实现。以其中准备好的复合物结构1tvk_prep.pdb为例:
1 2 3 4 5 6 7 | pyflare dynamics.py 1TVK_prep.pdb \ -C protein,water,ligand \ -f open --open-version 2.2.1 \ -s 0 \ -w explicit \ -u EP \ -d 1TVK |
其中:
- C选项:告诉Dynamics将复合物结构中的蛋白、配体与水链纳入计算
- 蛋白力场:用默认的AMBER ff14SB
- f选项:定义Dynamics的配体力场参数为Open Force Field
- open-version选项:定义OpenFF的版本为2.2.1
- 配体电荷:用默认的AM1-BCC
- w选项:定义使用显式水模型
- u选项:告诉Dynamics体系中的配体名称为EP
- 溶剂化水模型:TIP3P,随后加入 Na⁺ 和/或 Cl⁻ 离子以使整个系统呈电中性
- s选项:为0,定义Dynamics不跑MD而仅输出体系的原子坐标与拓扑文件。
- d选项:输出目录。
处理完毕,可在输出目录(1TVK)发现两个文件:
1 2 3 | ├── 1TVK ├── 1TVK_Dynamics.inpcrd #即所需的坐标文件(rst7) └── 1TVK_Dynamics.prmtop #即所需的拓扑文件(prm7) |
结构的初始化
OpenBPMD所有模拟均使用 OpenMM 分子动力学引擎7-9。系统平衡过程包括:首先进行最多 10,000 步(或能量收敛至 10 kJ/mol 的容差内)的能量最小化;随后在 NVT 系综下进行 500 ps 的带限制条件的平衡模拟,时间步长为 2 fs。在平衡过程中,非水重原子被限制在初始坐标位置上,力常数为 5 kcal/mol/Å2。所有生产模拟均在 NVT 系综下进行,使用 Langevin 积分器,时间步长为 4 fs,并采用了氢质量再分配方法(将氢原子质量设为 4 Da)。热浴参比温度设为 300 K,热浴耦合摩擦系数为 1 ps-1。模拟中应用了周期性边界条件,并采用粒子网格Ewald(PME)方法处理长程静电相互作用,截断半径为 10.0 Å。
增强采样模拟
OpenBPMD所选的集体变量(CV)是配体重原子的均方根偏差(RMSD),参比结构为平衡阶段结束时的坐标。该CV还结合了锚定原子(用于对齐蛋白质,使得无论蛋白质如何移动,RMSD值始终在相同的参考系下计算)以及配体的重原子。锚定原子的选择依据Clark等人3所述的标准进行。为了修正OpenMM在计算模拟过程中配体RMSD时未考虑周期性边界条件的问题,在锚定原子与配体之间施加了一个无力常数的平底约束势(flat-bottom restraint)。
高斯势垒的高度(hill height)是元动力学中最重要的参数之一。在Lukauskis等人6的研究中使用0.3和0.05 kcal/mol两种不同的势垒高度测试该参数对结合模式稳定性的影响,对整个数据集分别运行了两次模拟。例外的是DPP4体系,该体系全程仅使用了0.05 kcal/mol的势垒高度,以与Clark等人3设置保持一致。
在RMSD集体变量(CV)上应用了高斯宽度为0.002 nm的偏置势,每100 ps沉积一次偏置势,偏置因子(bias factor)为4。所有OpenBPMD模拟均运行了10 ns。报告的OpenBPMD打分是10次独立元动力学模拟所得打分的平均值。
打分函数
Clark等人3在评估配体结合模式的稳定性时使用了两个打分指标:PoseScore 和 PersScore。PoseScore 通过计算配体重原子的均方根偏差(RMSD)来评估构象稳定性,计算前会先根据蛋白质二级结构的 Cα 原子对模拟轨迹进行对齐。该指标在处理配体骨架发生显著位移的情况时表现尤为高效。然而,它难以检测到配体的微小移动,即使这些微小变化可能足以破坏蛋白质与配体之间的相互作用网络。因此,引入了 PersScore 这一指标,用于监测在元动力学模拟过程中持续存在的氢键比例,从而评估构象的稳定性。然而,PersScore 仅关注氢键,而无法反映除氢键以外的其他原子间相互作用,这导致对于那些不与蛋白质形成氢键的配体,PersScore 无法提供有效的打分。
为了改进 PersScore,OpenBPMD设计了一种新的打分指标,用于在BPMD模拟过程中追踪配体与蛋白质之间非键相互作用的持续性,称为 ContactScore。与仅依赖氢键的 PersScore 不同,ContactScore 基于一个更通用的“接触”定义:只要配体或蛋白质中的两个重原子之间的距离在 3.5 Å 以内,即视为形成一次接触。这样一来,就不再区分不同类型的非共价相互作用,例如 π–π 堆积、π–卤素作用或氢键等。在具体操作中,每 100 ps 记录一次接触的数量,并与 OpenBPMD 模拟开始时的接触数量进行比较。最终的 ContactScore 是通过对模拟最后 2 ns 内的接触数量取平均值得到的。
为了融合原有打分体系与新指标的优势,OpenBPMD使用了一个名为 CompScore 的组合打分,定义如下(eq 1):
$$
CompScore = PoseScore − 5 \times ContactScore \cdots(1)
$$
该公式源自Clark等人3补充材料中提供的公式3,在OpenBPMD实现中用前述的 ContactScore 替换了 PersScore。PoseScore 和 ContactScore 均使用 MDAnalysis10,11进行计算。模拟轨迹通过 MDTraj 12 进行后处理,以将溶质置于模拟盒子中心,并考虑周期性边界条件的影响。上述工作流程总结如图 1 所示。
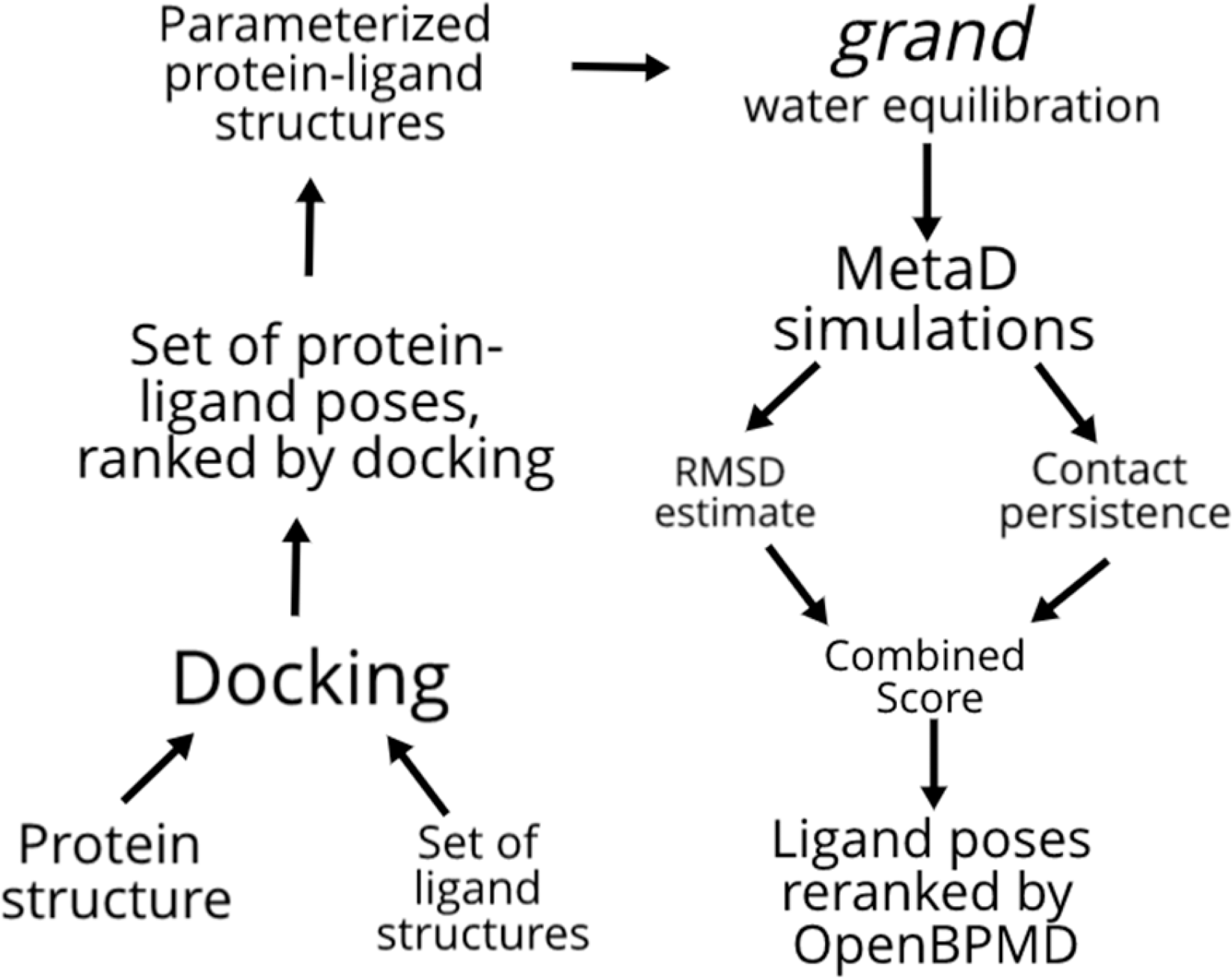
图1. OpenBPMD工作流,图片来自文献6。 工作流的左侧部分可以选用任何分子对接程序完成,而蛋白质-配体结构可以使用任何力场进行参数化。
图1展示了OpenBPMD的工作流程,始于一个蛋白质结构和一组配体结构。这些结构进行对接计算,得到一组由对接打分排序的蛋白质-配体结合模式(poses)。然后对这些结合模式进行参数化,并进行GRAND水合平衡(grand water equilibration),随后进行元动力学(MetaD)模拟。MetaD模拟产生RMSD估算值和接触持续性数据,这些数据综合成一个组合打分(Combined Score),该打分可用于配体结合模式的重新排序。需要指出的是,如果你是Flare的用户,你可以直接用Flare的GCNCMC来进行水合平衡,非常便利。
用于复现本文中展示结果的脚本openbpmd.py可从github数据仓库获得。就1TKV体系而言,重复10次计算的命令如下:
1 2 3 4 5 6 7 | pyflare openbpmd.py \ -s 1TVK_Dynamics.inpcrd \ -p 1TVK_Dynamics.prmtop \ -o ligand_pose0 \ -lig_resname EP \ -nreps 10 -hill_height 0.300 |
用同样的方式,完成PDB 4I50体系的BPMD计算。所需输入文件可从附件获得。
结果
EpoA与Alpha-1β微管蛋白的结合模式
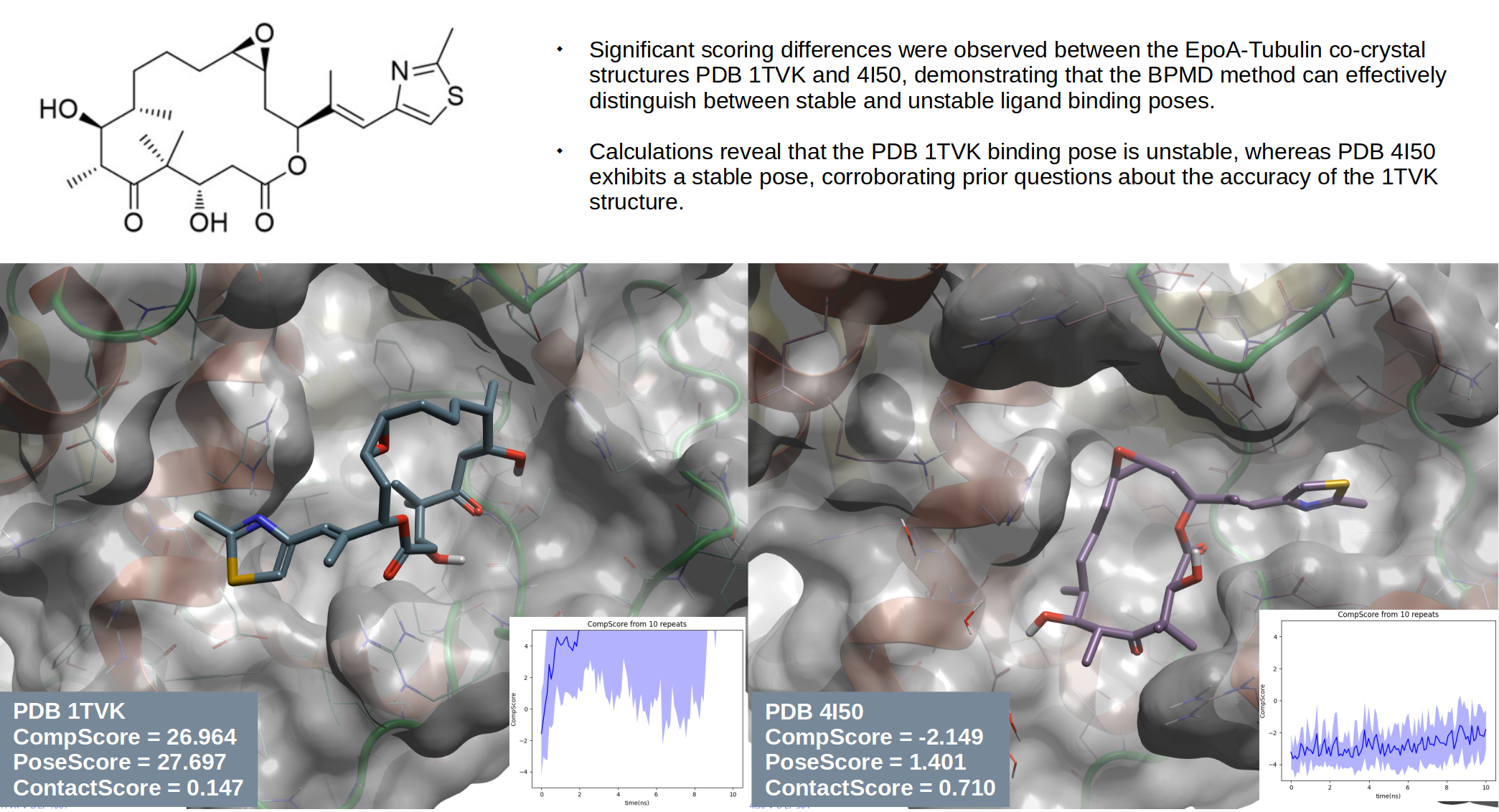
图2. EpoA的化学结构
天然产物Epothilones是一类能够结合到Alpha-1β-微管蛋白的公共有结合位点上稳定微管的抗真菌剂。2004年,Nettles等人15通过电子晶体学、核磁共振(NMR)和分子建模相结合的方法,首次解析了EpoA(图2)与α,β-微管蛋白结合的原子模型(PDB 1TVK,见图3-左)。然而,由于该模型中提出的EpoA结合模式与核磁共振数据存在不一致之处,因此受到了严重质疑。2013年,Prota等人16报道了一个分辨率为2.3 Å的EpoA与Alpha-1β-微管蛋白、类组蛋白蛋白RB3以及微管酪氨酸连接酶复合物的X-衍射晶体结构(PDB 4I50),与1TVK模型相比,EpoA在此结构中呈现出不同的结合模式(图3-右)。基于上述原因,在Fusani等人4的研究中采用了EpoA与微管蛋白α-1β链结合的结构来检验BPMD方法区分正确与错误配体结合模式的能力。在本文中,也以这个体系为例来演示Flare与OpenPBMD来实现同样的计算。
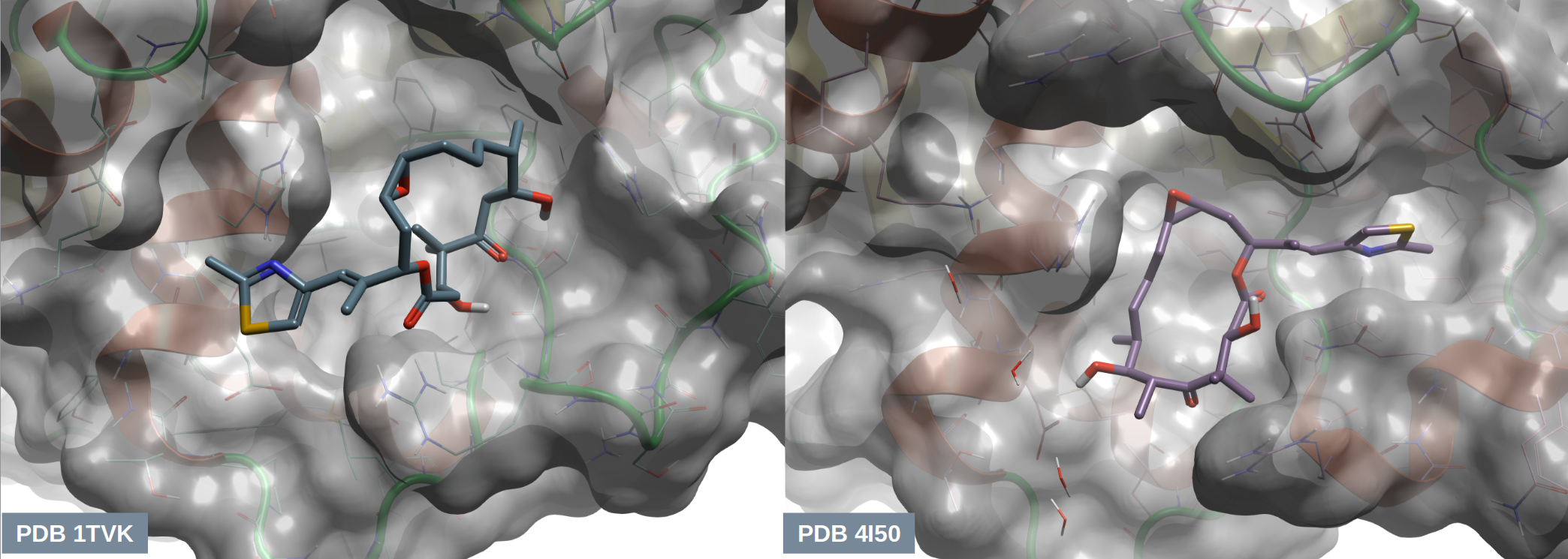
图3. EpoA与Alpha-1β-微管蛋白的共晶结构,左:PDB 1TVK;右:PDB 4I50。棍状分子:EpoA;分子表面:Alpha-1β-微管蛋白。图片用Flare V10生成。
用BPMD来评估EpoA的结合模式
计算完毕,在ligand_pose0目录下会生成10次计算的打分值统计结果results.csv。PDB 1TVK与4I50重复10次OpenBPMD计算的各个打分平均值与变异系数如表1所示。
表1. PDB 1TVK与4I50的10次重复OpenBPMD计算打分值的平均值与变异系数
| PDB Code | CompScore | CompScoreSD | PoseScore | PoseScoreSD | ContactScore | ContactScoreSD |
|---|---|---|---|---|---|---|
| 1TVK | 26.964 | 22.481 | 27.697 | 21.753 | 0.147 | 0.327 |
| 4I50 | -2.149 | 1.623 | 1.401 | 0.784 | 0.710 | 0.278 |
各个打分值解释如下:
- CompScore:综合了结合模式稳定性(PoseScore)和接触持久性(ContactScore)的最终打分。数值越低,表示结合模式越稳定(PoseScore 低 + ContactScore高)。用于同一个化合物不同结合模式的比较,优先选择CompScore最低的结合模式。
- CompScoreSD:综合打分标准差。多次模拟中CompScore的标准差,反映结果的波动性。数值高,表明不同重复模拟间的CompScore差异大,结果可靠性较低。数值低,表明结果稳定可靠。
- PoseScore:结合模式稳定性打分,配体在模拟过程中相对于初始姿势的RMSD平均值(单位:Å)。数值越低,表示配体在模拟中越少偏离初始结合模式,越稳定。若PoseScore \(<\) 2.0 Å,通常认为结合模式正确,接近天然结合模式。
- PoseScoreSD:结合模式稳定性标准差,即多次模拟中PoseScore的标准差。数值高,表明配体在模拟中可能发生较大位移,比如如部分模拟中配体脱离结合位点。需结合ContactScore判断是否因结合模式不稳定或模拟参数不当导致。
- ContactScore:接触持久性打分,模拟中配体与蛋白间原子接触的持久性(0~1),模拟最后2 ns的接触数平均值 ÷ 初始接触数。数值越高(接近1),表明配体与蛋白的接触保持良好。若ContactScore \(<\) 0.5,可能提示初始姿势存在关键接触缺失。
- ContactScoreSD:接触持久性标准差,多次模拟中ContactScore的标准差。数值高,表明接触数在不同模拟中波动大,可能因水分子或蛋白构象变化导致。需检查溶剂环境,比如是否使用GCMC优化水分子。
如表1所示,在对初始结构PDB 1TVK进行元动力学计算的过程中:CompScore = 26.964,PoseScore = 27.697,ContactScore = 0.147。这说明了:
- 结合模式稳定性差:PoseScore = 26.964 Å远高于2.0 Å,这意味着配体在模拟中大幅偏离初始结合模式。
- 接触持久性一般:ContactScore = 0.147,仅保留约14.7%的初始接触。
- 结果可靠性低:CompScoreSD = 22.481和PoseScoreSD = 21.753表明多次模拟差异极大,需要检查:(1)是否初始姿势存在明显错误?(2)是否未使用GCMC优化水分子?(3)是否需要增加模拟次数或调整元动力学参数(如降低山高度)?
在对初始结构PDB 4I50进行元动力学计算的过程中:CompScore = -2.149,PoseScore = 1.401,ContactScore = 0.710。这说明了:
- 结合模式稳定性很好:PoseScore = 1.401 Å 低于2.0 Å,这意味着配体在模拟中几乎不偏离初始结合模式。
- 接触持久性很好:ContactScore = 0.710,持续保留约71%的初始接触。
- 结果可靠性极高:CompScoreSD = 1.623和PoseScoreSD = 0.7843表明多次模拟差异极小。
总的来说,从打分函数CompScore上看,可以得到两个结论:
- 在原始结构(PDB 1TVK)与较新解析结构(PDB 4I50)的BPMD打分之间存在显著差异,这说明BPMD方法能够有效区分稳定与不稳定的配体结合模式。
- BPMD打分表明PDB 1TVK的结合模式是不稳定,而PDB 4I50的结合模式是稳定的,这与此前对PDB 1TVK结构的准确性质疑是一致的。
此外,从PoseScore与ContactScore上看,也得到同样的结果。
可视化分析BPMD的计算结果
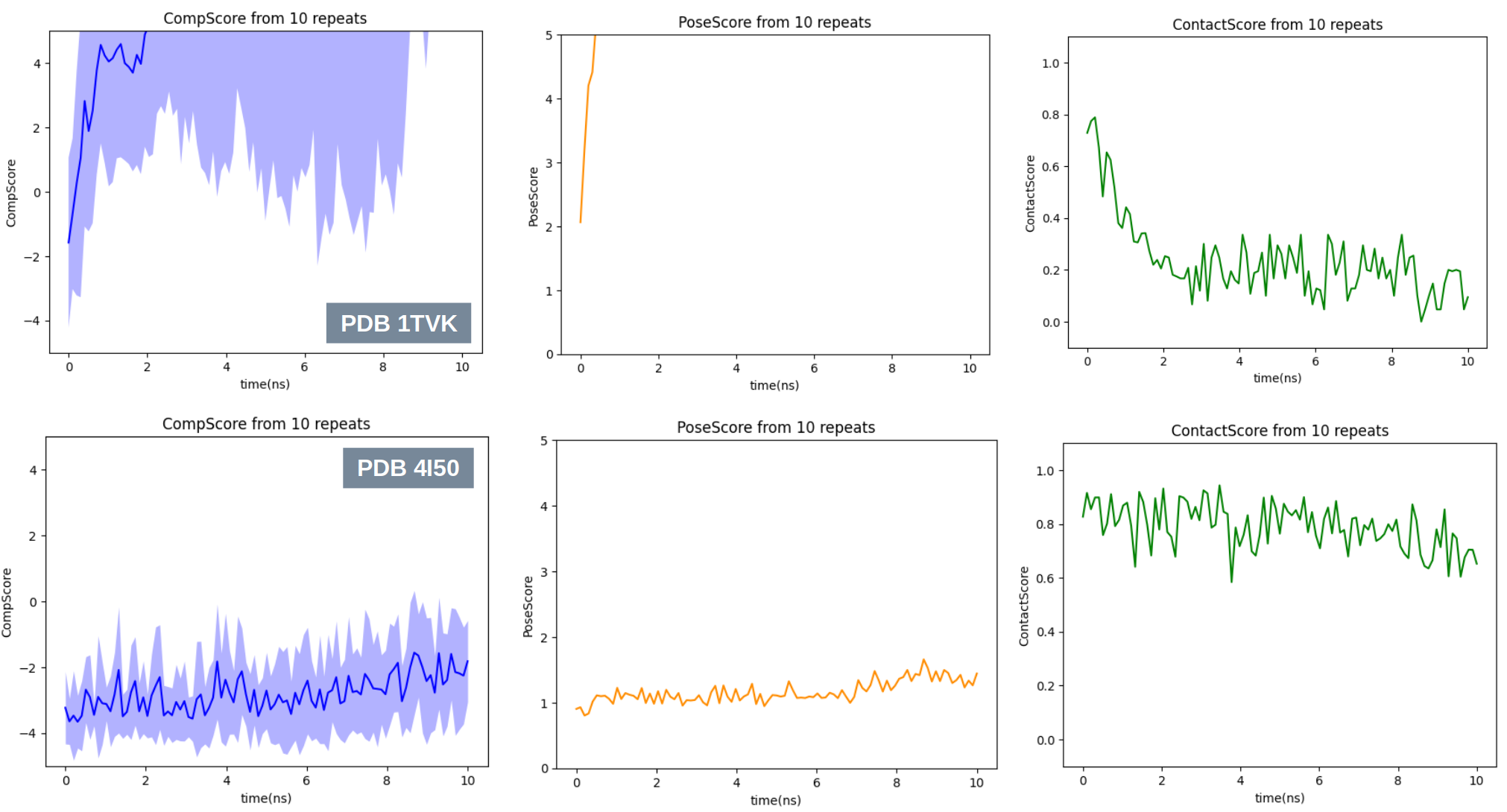
图4. EpoA与Alpha-1β-微管蛋白共晶结构进行10次BPMD模拟的平均打分值随着元动力学模拟进行的变化曲线。上:PDB 1TVK;下:PDB 4I50。图片用matplotlib软件包的pyplot绘制。
经过10次重复模拟后,绘制了打分均值随着元动力学模拟进行的变化曲线。在PDB 1TVK体系里(如图4-上所示),可以看到随着元动力学模拟的进行,与初始的结合模式相比,最后2纳秒内的CompScore与PoseScore打分高到看不见,说明EpoA在BPMD偏置下发了剧烈的重排;而ContactScore非常低,说明初始的相互作用得不到维持;BPMD打分说明该配体在1TVK中的结合模式是不稳定的。相反,在PDB 4I50体系里(如图4-下所示),随着元动力学模拟的进行,与初始的结合模式相比,EpoA在BPMD偏置下相当平稳,说明该配体在4I50中的结合模式是稳定的。两个体系的曲线差异极其显著,直观地表明OpenBPMD方法能够有效区分不同的结合模式。这一结果也此前对1TKV结构合理性地质疑相一致,进一步验证了方法的可靠性与性能。
结论
本文介绍了结合模式元动力学(Binding Pose MetaDynamics,BPMD)模拟的开源python实现OpenBPMD,演示了Flare与OpenBPMD联合应用可大幅降低计算门槛,为研究者提供了一个能快速构建研究体系并高效完成结合模式稳定性评估的完整工作流。
具体而言,本文以微管蛋白α-1β与Epothilone A(EpoA)的结合模式为例,展示了Flare与OpenBPMD联用在评估蛋白质-配体结合模式稳定性中的强大能力。结果表明,Flare以非常友好、易用的方式提供了MetaD模拟所需的输入文件,而OpenBPMD能够有效区分实验争议结构中稳定与不稳定的结合模式:对于早期因实验方法受限而被质疑的PDB 1TVK模型,其CompScore(26.964)和PoseScore(27.697 Å)显著偏离稳定阈值(PoseScore \(<\) 2.0 Å),且接触持续性(ContactScore = 0.147)极低;而分辨率高的结构PDB 4I50则表现出优异的稳定性(PoseScore = 1.401 Å,ContactScore = 0.710),充分验证了OpenBPMD打分与实验观测的一致性。这一发现不仅验证了Flare与OpenBPMD联用方法的可靠性,也为解决实验结构争议提供了计算生物学视角的佐证。近期OpenBPMD在Protac分子设计中的成功应用13,更彰显了该方法在复杂分子体系中的拓展潜力。
接下来可以作什么?
- 用OpenBPMD大规模评估分子对接结合模式的稳定性
- 用OpenBPMD大规模评估Spark结果在结合位点里的结合模式稳定性
- 在Flare FEP计算之前评估结合模式的构象稳定性,过滤掉不稳定的结合模式以节省计算资源17
- 可探索与基于AI结构预测联用的工作流,构建”生成-验证-优化”闭环系统,为基于结构的药物设计提供更全面的解决方案
附件
为了重现本文的结果,提供了原始的输入文件:
1 2 3 4 5 6 7 | . ├── 1TVK_Dynamics.inpcrd.gz ├── 1TVK_Dynamics.prmtop.gz ├── 1TVK_prep.pdb ├── 4I50_Dynamics.inpcrd.gz ├── 4I50_Dynamics.prmtop.gz └── 4I50_prep.pdb |
文件可从GitHub下载:pyflare_example
在药物设计过程中Flare全程陪你
Flare交付先进的科学方法、分析工具和直观、易用的增强功能,洞察您的配体-蛋白质复合物结构。
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出重要的分子。
- 电邮:info@molcalx.com
- 电话:020-38261356
文献
- Mey, A.S.J.S. et al. (2020) “Best Practices for Alchemical Free Energy Calculations [Article v1.0],” Living Journal of Computational Molecular Science, 2(1), pp. 1–48. Available at: https://doi.org/10.33011/livecoms.2.1.18378.
- Wang, Z. et al. (2016) “Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power,” Physical Chemistry Chemical Physics, 18(18), pp. 12964–12975. Available at: https://doi.org/10.1039/C6CP01555G.
- Clark, A.J. et al. (2016) “Prediction of Protein–Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations,” Journal of Chemical Theory and Computation, 12(6), pp. 2990–2998. Available at: https://doi.org/10.1021/acs.jctc.6b00201.
- Fusani, L. et al. (2020) “Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics,” Journal of Chemical Information and Modeling, 60(3), pp. 1528–1539. Available at: https://doi.org/10.1021/acs.jcim.9b00843.
- Miller, E.B. et al. (2021) “Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein–Ligand Binding,” Journal of Chemical Theory and Computation, 17(4), pp. 2630–2639. Available at: https://doi.org/10.1021/acs.jctc.1c00136.
- Lukauskis, D. et al. (2022) “Open Binding Pose Metadynamics: An Effective Approach for the Ranking of Protein–Ligand Binding Poses,” Journal of Chemical Information and Modeling, 62(23), pp. 6209–6216. Available at: https://doi.org/10.1021/acs.jcim.2c01142.
- Eastman, P. et al. (2013) “OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation,” Journal of Chemical Theory and Computation, 9(1), pp. 461–469. Available at: https://doi.org/10.1021/ct300857j.
- Eastman, P. et al. (2017) “OpenMM 7: Rapid development of high performance algorithms for molecular dynamics,” PLOS Computational Biology. Edited by R. Gentleman, 13(7), p. e1005659. Available at: https://doi.org/10.1371/journal.pcbi.1005659.
- Eastman, P. et al. (2024) “OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials,” The Journal of Physical Chemistry B, 128(1), pp. 109–116. Available at: https://doi.org/10.1021/acs.jpcb.3c06662.
- Michaud‐Agrawal, N. et al. (2011) “MDAnalysis: A toolkit for the analysis of molecular dynamics simulations,” Journal of Computational Chemistry, 32(10), pp. 2319–2327. Available at: https://doi.org/10.1002/jcc.21787.
- Gowers, R. et al. (2016) “MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations,” in Proceedings of the 15th Python in Science Conference, pp. 98–105. Available at: https://doi.org/10.25080/Majora-629e541a-00e.
- McGibbon, R.T. et al. (2015) “MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories,” Biophysical Journal, 109(8), pp. 1528–1532. Available at: https://doi.org/10.1016/j.bpj.2015.08.015.
- Ma, B. et al. (2024) “A Top-Down Design Approach for Generating a Peptide PROTAC Drug Targeting Androgen Receptor for Androgenetic Alopecia Therapy,” Journal of Medicinal Chemistry, 67(12), pp. 10336–10349. Available at: https://doi.org/10.1021/acs.jmedchem.4c00828.
- Flare™, version 10, Cresset®, Litlington, Cambridgeshire, UK; https://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130
- Nettles, J.H. et al. (2004) “The Binding Mode of Epothilone A on α,ß-Tubulin by Electron Crystallography,” Science, 305(5685), pp. 866–869. Available at: https://doi.org/10.1126/science.1099190.
- Prota, A.E. et al. (2013) “Molecular Mechanism of Action of Microtubule-Stabilizing Anticancer Agents,” Science, 339(6119), pp. 587–590. Available at: https://doi.org/10.1126/science.1230582.
- Heinzelmann, G. and Gilson, M.K. (2021) “Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation,” Scientific Reports, 11(1), p. 1116. Available at: https://doi.org/10.1038/s41598-020-80769-1.


